GxP Wörterbuch
A0-Value (A0 -Wert, Letalitätswert)
Die Norm EN DIN ISO 15883-1 definiert den A0-Wert als Maß für die mikrobielle Abtötung bei der Desinfektion mit feuchter Hitze. Dieser Wert wird verwendet, um die erforderliche Menge an feuchter Hitze in maschinellen Reinigungs-Desinfektions-Geräten festzulegen. Der A0-Wert repräsentiert die Zeit, die nötig ist, um eine bestimmte Log-Reduktion von Mikroorganismen basierend auf der Temperatur des Desinfektionszyklus zu erreichen.

ALCOA (++) Prinzip
Das ALCOA-Prinzip soll sicherstellen, dass Datenintegrität und Datenqualität in Aufzeichnungen gewahrt bleiben. ALCOA steht für:
- Attributable - Daten müssen eindeutig einer Quelle oder Person zuzuordnen sein.
- Legible - Daten müssen lesbar und dauerhaft sein.
- Contemporaneous - Daten sollen zum Zeitpunkt der Aktivität oder Beobachtung erfasst werden.
- Original - Daten sollten in ihrer ursprünglichen, unveränderten Form vorliegen.
- Accurate - Daten müssen korrekt und fehlerfrei sein.
Das ALCOA++ Prinzip erweitert das ursprüngliche ALCOA-Konzept, um zusätzliche Attribute der Datenintegrität in regulierten Umgebungen wie dem GxP-Umfeld zu berücksichtigen. Diese Erweiterung umfasst weitere Kriterien:
- Complete - Alle Daten müssen vollständig und umfassend sein.
- Consistent - Daten sollten in einem zuverlässigen und logischen Format vorliegen.
- Enduring - Daten müssen dauerhaft erhalten bleiben.
- Available - Daten sollten jederzeit zugänglich sein, wenn sie benötigt werden.
Annex 1
Seit August 2023 ist der neue Annex 1 in einer überarbeiteten Fassung gültig, mit einer Übergangsfrist bis 2024. Obwohl der grundsätzliche Aufbau gleichgeblieben ist, hat die Ergänzung durch weitere Unterkapitel den Annex umfangreicher und detaillierter gemacht. Der Fokus liegt besonders auf der Herstellung steriler Produkte, um das Risiko von mikrobiologischen, partikulären und pyrogenen Kontaminationen zu minimieren. Zudem dient der neue Annex 1 als eine Handlungsanleitung für den bestmöglichen Schutz steriler Produkte.
API – Active Pharmaceutical Ingredient (Wirkstoff)
Ein Active Pharmaceutical Ingredient (API) ist der pharmakologische Hauptwirkstoff eines Arzneimittels, der die beabsichtigte therapeutische Wirkung erzielt. APIs sind hochreine chemische Verbindungen, die in strikte Herstellungsprozesse eingebunden sind, um ihre Qualität, Sicherheit und Wirksamkeit sicherzustellen. Sie werden in Medikamenten häufig mit Hilfsstoffen kombiniert, um die Darreichungsform und Stabilität des Endprodukts zu verbessern.
APR – Annual Product Review (Jährliche Produktüberprüfung)
Die von der FDA geforderte Annual Product Review (APR) ist ein obligatorischer Prozess zur Sicherstellung der Qualität von pharmazeutischen Produkten, die in den USA hergestellt oder importiert werden. Der APR umfasst eine retrospektive, systematische Überprüfung aller Chargen, Produktionsprozesse, Abweichungen, Reklamationen sowie jeglicher Änderungen in Herstellungsverfahren und Kontrollmethoden innerhalb eines Jahres.
Audit
Im GxP-Umfeld bezieht sich ein Audit auf eine systematische und unabhängige Untersuchung vor Ort, die durchgeführt wird, um sicherzustellen, dass alle Prozesse, Systeme und Verfahren konform mit den geltenden Vorschriften und Qualitätsstandards sind. Audits dienen dazu, die Integrität und Qualität von Produkten zu gewährleisten, indem sie Schwachstellen und Risiken identifizieren sowie die Effektivität von Qualitätskontrollmaßnahmen bewerten. Audits können von unterschiedlichen Stellen, sowohl intern als auch extern, beispielsweise durch Kunden, durchgeführt werden. Begrifflich zu unterscheiden, jedoch mit demselben Hintergrund, ist die Inspektion, die ausschließlich durch die zuständigen Aufsichtsbehörden durchgeführt werden darf.
Audit Trail
Als Audit Trail bezeichnet man eine geordnete Aufzeichnung aller Änderungen und Aktionen innerhalb eines Systems oder Dokuments. Diese Protokollierung stellt sicher, dass die Datenintegrität und Rückverfolgbarkeit gewährleistet sind, was entscheidend für die Einhaltung regulatorischer Standards ist. Der Audit Trail hilft, nachvollziehbar zu machen, wer, wann und warum Änderungen vorgenommen hat, und ist ein wichtiges Instrument der Qualitätssicherung. Im Zuge der Digitalisierung hat der Begriff auch in Bezug auf Datensicherheit und Datenintegrität an Bedeutung gefunden und wird in Regularien wie 21 CFR part11, ALCOA++ oder EU GMP Annex 11 explizit erwähnt.
Bioburden (Keimbelastung)
Beschreiben die Gesamtzahl aller lebensfähigen Mikroorganismen, die auf einer nicht-sterilisierten Oberfläche oder im Produkt vorhanden sind.
BP – British Pharmacopoeia (Arzneibuch UK)
Die British Pharmacopoeia (BP) ist das offizielle Arzneibuch des Vereinigten Königreichs und bietet umfassende Standards zur Sicherstellung der Qualität und Sicherheit von Arzneimitteln. Sie dient als wichtiges Referenzwerk für Hersteller, Prüflabore und Aufsichtsbehörden.
21 CFR – Code of Federal Regulations
Der Code of Federal Regulations (CFR) ist eine systematische Sammlung der allgemeinen und dauerhaften Vorschriften, die von den Ministerien und Behörden der US-Bundesregierung im Federal Register veröffentlicht werden. Titel 21 des CFR widmet sich speziell den Vorschriften der Food and Drug Administration (FDA) und behandelt Aspekte zur Regelung von Lebensmitteln und Arzneimitteln.
21 CFR part11
21 CFR-Part 11 ist eine Regelung der U.S. Food and Drug Administration (FDA), die die Kriterien für die Annahme elektronischer Aufzeichnungen und elektronischer Signaturen regelt. Sie legt die Anforderungen an die Integrität, Sicherheit und Verfügbarkeit dieser Aufzeichnungen fest, um sicherzustellen, dass sie ebenso vertrauenswürdig und zuverlässig sind wie traditionelle papierbasierte Dokumente.
Calibration (Kalibrierung)
Kalibrierung ist der dokumentierte Vergleich des zu kalibrierenden Messgeräts mit einer rückführbaren Referenz.
Alle Sensoren und Messgeräte müssen vor der Verwendung für eine Validierungsstudie kalibriert werden. Kalibrierungsergebnisse müssen dokumentiert werden. Erreichbare Messunsicherheiten müssen in Einklang mit den anwendungsspezifischen Anforderungen stehen.
- Nach ISO/IEC GUIDE 99:2007 ist die Kalibrierung ein Vorgang, der eine Beziehung zwischen den Messgrößen und entsprechenden Angaben herstellt
- Kalibrierung = ermittelte Differenz zwischen dem gemessenen Wert und dem wahren Wert (rückverfolgbare Referenz/Standard)
- Justage = Korrektur des gemessenen Werts hin zum wahren Wert, um Messfehler zu minimieren und die Genauigkeit zu erhöhen
- Kalibrierung & Justage erfolgt während der gesamten Lebensdauer eines Systems, Instruments, Sensors und muss durchgeführt werden.
- Sie ist Teil der Qualifizierung und der Validierung, ersetzt diese aber nicht.
- Sie umfasst alle Sensoren und Messgrößen, Instrumenten oder einem System und definieren die Gesamtgenauigkeit der gesamten Messkette.
- Sie bezieht sich auf die Leistung und Eigenschaften von Sensoren, Messsystemen und Software
- Sie schließt allgemeine Methoden zur Metrologie ein:
- Die Definition des Messverfahrens (Umgebungsbedingungen; erforderliche Standards Method).
- Erstellung eines mathematischen Modells zur Bewertung der Kalibrierung einschließlich ihrer
- Durchführung der Kalibrierung
- Erstellung eines Kalibrierzertifikat mit Details (Ermittelte Abweichung, Justage und Messunsicherheit).
- Abhängig vom Prozess/ Sensor/ Messgröße ist eine Nachkalibrierung ohne Justage zu Überprüfungs- und Dokumentationszwecken nach dem Gebrauch erforderlich.
Calibration Protocol (Kalibrierungsprotokoll)
Ein Kalibrierprotokoll ist ein technisches Dokument, das einen umfassenden Plan oder Standardarbeitsanweisung beschreibt, wie die Kalibrierung eines Geräts, Instruments oder Systems durchgeführt werden soll. Es enthält alle notwendigen Schritte und Bedingungen, um sicherzustellen, dass das Gerät präzise Messungen liefert und innerhalb der spezifizierten Toleranzen arbeitet. Ein Kalibrierprotokoll legt spezifische Kalibrieranforderungen fest, einschließlich der zu verwendenden Methoden, der Kalibrierintervalle, der akzeptablen Grenzwerte und der erforderlichen Dokumentation der Ergebnisse.
CAPA – Corrective and Preventive Action (Korrektur- und Vorbeugungsmaßnahmen)
Ist ein systematischer Ansatz zur Identifizierung und Behebung von Ursachen für Abweichungen sowie zur Implementierung präventiver Maßnahmen, um deren Wiederholung zu verhindern. Ein robustes CAPA-System beinhaltet die Ursachenanalyse, die Implementierung von Korrekturmaßnahmen, die Evaluation der Effektivität dieser Maßnahmen und selbstverständlich die lückenlose begleitende Dokumentation.
CC – Change Control (Änderungskontrolle)
Die Änderungskontrolle ist ein systematischer und formaler Prozess, der sicherstellt, dass alle Änderungen an Produkten, Prozessen oder Systemen sorgfältig geplant, bewertet und dokumentiert werden. Das Hauptziel der Änderungskontrolle im GMP-Umfeld besteht darin, die Integrität und Qualität von Produkten trotz Änderungen zu wahren und sicherzustellen, dass diese Änderungen die regulatorischen Anforderungen erfüllen.
CFR – Code Federal Regulations (Bundesrichtlinien der USA)
Sammlung der allgemeinen und permanenten Regeln, die von den Bundesbehörden der USA herausgegeben werden. Im Bereich der pharmazeutischen und biotechnologischen Industrie spielt insbesondere der 21 CFR eine zentrale Rolle, da er die Vorgaben der U.S. Food and Drug Administration (FDA) zur Gewährleistung der Sicherheit, Wirksamkeit und Qualität von pharmazeutischen Produkten und medizinischen Geräten enthält.
CFU – Colony Forming Unit (Koloniebildende Einheit KBE)
CFU ist eine Maßeinheit, die in der Mikrobiologie verwendet wird, um die Anzahl lebender Mikroorganismen in einer Probe zu schätzen. CFUs sind entscheidend für die Bewertung der mikrobiologischen Kontamination von Produkten und Materialien. Eine Koloniebildende Einheit wird definiert als vermehrungsfähige Mikroorganismen, wie zum Beispiel Bakterien oder Pilze, die während der Kultivierung zur Bildung mehrerer Kolonien führen.
ChP – Chinese Pharmacopoeia (Arzneibuch China)
Das chinesische Arzneibuch (ChP), auch bekannt als PPRC, ist das offizielle Kompendium der Volksrepublik China und enthält umfassende Standards für die Reinheit, Beschreibung, Tests, Dosierung, Sicherheitsvorkehrungen, Lagerung und Stärke von Arzneimitteln. Es spielt eine entscheidende Rolle bei der Sicherstellung der Arzneimittelqualität in China, ähnlich wie das Europäische Arzneibuch in Europa und die USP in den USA.
CIP – Clean in Place (Ortsgebundene Reinigung)
Dies ist ein Verfahren zur Reinigung von Produktionsanlagen und -systemen ohne deren Demontage. CIP wird in der Pharmaindustrie eingesetzt zur Reinigung von z.B. Rohren, Behältern und Abfüllanlagen oder Tanks. Das Verfahren verwendet eine Kombination aus Spülen, Reinigen und Desinfizieren mit verschiedenen Reinigungsmitteln. Oftmals erfolgt im Anschluss die Sterilisation über die sogenannte SIP (Steam in Place).
Cold Chain (Kühlkette)
Eine Kühlkette ist ein temperaturkontrollierter Lieferprozess, bei dem temperaturempfindliche Produkte wie Lebensmittel, Arzneimittel, chemische Substanzen und biologische Materialien über ihre gesamte Lieferkette hinweg innerhalb eines bestimmten Temperaturbereichs transportiert und gelagert werden. Ziel ist es, die Qualität, Sicherheit und Wirksamkeit dieser Produkte zu gewährleisten, indem die vorgegebenen Temperaturanforderungen strikt eingehalten werden. Die Kühlkette umfasst den gesamten Prozess, beginnend bei der Herstellung über Lagerung, Transport und Verteilung bis hin zum Endverbraucher.
Im Bereich Pharma handelt es sich dabei u.a. um Arzneimittel, Wirkstoffe (APIs), Impfstoffe und biologische Substanzen. Diese Produkte reagieren äußerst empfindlich auf Temperaturschwankungen, und jede Abweichung vom empfohlenen Bereich kann sie unwirksam oder sogar schädlich machen.
Cold Chain Management (Kühlketten-Management)
Cold Chain Management bezeichnet die umfassende Steuerung und Überwachung aller Phasen innerhalb einer temperaturkontrollierten Lieferkette, um die Integrität von temperaturempfindlichen Produkten wie Lebensmitteln, pharmazeutischen Produkten und biologischen Materialien zu gewährleisten. Dies umfasst den gesamten Prozess von der Herstellung über Lagerung und Transport bis hin zur endgültigen Übergabe an den Endverbraucher. Durch effizientes Cold Chain Management wird sichergestellt, dass diese Produkte wirksam und sicher bleiben sowie alle relevanten regulatorischen Anforderungen erfüllt werden.
Compliance (Einhaltung von Vorschriften)
Compliance im regulierten Umfeld bezieht sich auf die Einhaltung aller relevanten gesetzlichen, ethischen und regulatorischen Anforderungen und Standards. In Branchen wie der Pharma-, Lebensmittel- und Medizintechnikindustrie bedeutet dies, dass alle Prozesse, Systeme und Produkte den festgelegten Vorschriften und Best Practices entsprechen, um Sicherheit, Wirksamkeit und Qualität zu gewährleisten. Letztendlich geht es um die Sicherstellung der Patientensicherheit.
Concurrent Validation (Begleitende Validierung)
Die begleitende Validierung findet parallel zur laufenden Routineproduktion statt. Sie dient dazu, die Leistungsfähigkeit und Konsistenz des Herstellungsprozesses zu bewerten und sicherzustellen, dass kontinuierlich Qualitäts- und regulatorische Anforderungen eingehalten werden.
CPV – Continued Process Validation (Fortlaufende Validierung)
Die fortlaufende Prozessüberwachung (Continued Process Verification, CPV) stellt sicher, dass Produktionsprozesse und Komponenten stets innerhalb festgelegter Qualitätsgrenzen bleiben, und wird als dritte Phase einer Prozessvalidierung betrachtet. CPV zielt darauf ab, Prozesse kontinuierlich unter Kontrolle zu halten und Qualitätsstandards durch regelmäßige Überwachung zu gewährleisten. Eine effiziente CPV identifiziert Prozessinkonsistenzen und ermöglicht schnelle Korrekturmaßnahmen.
CQA – Critical Quality Attributes (Kritische Qualitätsmerkmale)
Kritische Qualitätsmerkmale (CQA) sind spezifische physikalische, chemische, biologische oder mikrobiologische Eigenschaften oder Merkmale eines Produkts, die innerhalb festgelegter Grenzwerte, Bereiche oder Verteilungen liegen müssen, um sicherzustellen, dass die gewünschte Produktqualität erreicht wird. Diese Merkmale sind entscheidend für die Herstellung und Sicherstellung eines konsistenten Qualitätsniveaus bei Produkten.
CSV – Computer-System/ -Software-Validation
The description refers to Computer System Validation (CSV), which is a critical process to ensure that a computer system or software performs as intended and operates error-free, especially in environments subject to current Good Manufacturing Practice (cGMP) guidelines. The CSV process includes requirements such as risk analysis, planning, testing and validation, documentation, and continuous monitoring. Proper validation of computerized systems is essential in maintaining quality assurance and compliance.
D-Value (D-Wert)
Der D-Wert, oder Dezimalreduktionszeit, ist die benötigte Zeit in Minuten, um die Keimzahl eines Mikroorganismus um 90% zu reduzieren, entsprechend einer log-Zyklus-Abtötung. Bei der Dampfsterilisation misst der D-Wert die Abtötungseffizienz des Prozesses. Er ist entscheidend für die Entwicklung und Validierung von Sterilisationsverfahren, da er sicherstellt, dass die mikrobiologische Belastung reduziert wird, ohne die Produktintegrität zu beeinträchtigen.
Data Integrity (Datenintegrität)
Datenintegrität bezieht sich auf die Vollständigkeit, Korrektheit, Konsistenz und Genauigkeit von Daten über deren gesamten Lebenszyklus. Es ist entscheidend für die Zuverlässigkeit der Daten, die in regulierten Industrien wie der Pharma- und Medizintechnik verwendet werden. Data Integrity gewährleistet, dass Daten vertrauenswürdig und nachvollziehbar sind, was wichtig für die Einhaltung regulatorischer Vorschriften und für die Sicherheit und Wirksamkeit von Produkten ist. Sie ist ein essenzieller Bestandteil des Qualitätssicherungssystem und wird in vielen Richtlinien und Normen als Grundlage einer normenkonformen Qualitätssicherung gefordert.
Design Specifications
Design Specifications bezieht sich auf detaillierte technische Dokumente, die die Anforderungen und Merkmale eines Systems oder Produkts beschreiben. Sie legen fest, wie ein System oder eine Komponente funktionieren soll, um bestimmte Standards und regulatorische Anforderungen zu erfüllen. Diese Spezifikationen bilden die Basis für die Entwicklung und Validierung von Systemen, wobei sie sicherstellen, dass alle relevanten Aspekte wie Funktionalität, Sicherheit und Compliance berücksichtigt werden.
Deviation (Abweichung)
Im pharmatechnischen Umfeld bezeichnet der Begriff "Abweichung" (englisch: "Deviation") eine Abweichung von den festgelegten Standards, Verfahren oder Spezifikationen innerhalb eines regulierten Prozesses, wie beispielsweise der Herstellung, Prüfung oder Qualitätskontrolle von Arzneimitteln. Abweichungen können auf Prozessfehler, menschliches Versagen oder unvorhergesehene Ereignisse zurückzuführen sein und müssen gründlich und detailliert dokumentiert, untersucht und entsprechend korrigiert werden, um die Sicherheit und Wirksamkeit der Produkte sicherzustellen.
Deviation Management (Abweichungsmanagement)
Als Abweichungsmanagement bezeichnet man den strukturierten Prozess zur Identifizierung, Dokumentation, Untersuchung und Behebung von Abweichungen, die während der Herstellung, Prüfung oder Qualitätskontrolle von Arzneimitteln auftreten. Dieser Prozess ist entscheidend, um die Integrität und Qualität der Produkte sicherzustellen und die Einhaltung regulatorischer Anforderungen zu gewährleisten.
Das Abweichungsmanagement umfasst die folgenden Schritte:
- Erkennung: Identifizierung einer Abweichung vom Standardverfahren oder der Spezifikation.
- Dokumentation: Schriftliche Aufzeichnung der Abweichung, einschließlich ihrer Details und möglichen Auswirkungen.
- Untersuchung: Analyse der Ursachen der Abweichung, um den Ursprung und die Gründe zu ermitteln.
- Korrekturmaßnahmen: Implementierung von Maßnahmen, um die unmittelbaren Auswirkungen der Abweichung zu beheben.
- Vorbeugende Maßnahmen: Einführen von Maßnahmen, um ein erneutes Auftreten der Abweichung zu verhindern.
- Überprüfung und Abschluss: Bewertung der Effektivität der ergriffenen Maßnahmen und formeller Abschluss der Abweichung.
Ein effektives Abweichungsmanagement ist zur Einhaltung regulatorischer Compliance und der kontinuierlichen Verbesserung der Produktqualität ausschlaggebend.
DMS – Document Management System (Dokumentenmanagementsystem)
Ein Dokumentenmanagementsystem (DMS) ist eine Softwarelösung, die den Umgang mit elektronischen Dokumenten innerhalb eines Unternehmens organisiert, speichert und verwaltet. Es ermöglicht Benutzern, Dokumente effizient zu erstellen, bearbeiten, speichern und abrufen, während es gleichzeitig Zugriffsrechte und Bearbeitungsprotokolle überwacht. In einer großen Organisation hilft ein DMS dabei, den Informationsfluss zu optimieren, Compliance-Anforderungen zu erfüllen und die Zusammenarbeit zwischen verschiedenen Geschäftsbereichen zu verbessern. Es kann sowohl strukturierte Daten wie Berichte und Richtlinien, als auch unstrukturierte Inhalte, wie E-Mails und Multimedia, in einer zentralen, zugänglichen Plattform verwalten.
DQ – Design Qualification (Design Qualifizierung)
Die DQ (Design Qualification) ist der dokumentierte Nachweis, dass die erstellten Planungsunterlagen den Benutzeranforderungen im Lastenheft entsprechen. Sie bestätigt zudem, dass alle erforderlichen Planungsunterlagen für die Umsetzung erstellt wurden und die GMP-Anforderungen (Gesetze, Richtlinien, Stand der Technik) berücksichtigt sind.
EMA – European Medicines Agency (Europäische Arzneimittel-Agentur)
Die EMA, oder European Medicines Agency, ist eine Agentur der Europäischen Union, die für die wissenschaftliche Bewertung, Überwachung und Sicherheit von Arzneimitteln in der EU verantwortlich ist. Sie spielt eine zentrale Rolle bei der Zulassung neuer Medikamente, um sicherzustellen, dass diese den hohen Qualitäts-, Sicherheits- und Wirksamkeitsstandards entsprechen und schützt und fördert somit die Gesundheit von Menschen und Tieren durch die Bewertung und Überwachung von Arzneimitteln innerhalb der Europäischen Union (EU) und des Europäischen Wirtschaftsraums (EWR).
EP – oder Ph.Eur European Pharmacopeia (Europäisches Arzneibuch)
Das Europäische Arzneibuch (Ph. Eur.) ist die Hauptquelle für offizielle Qualitätsstandards für Arzneimittel und deren Inhaltsstoffe in Europa. Die Europäische Direktion für die Qualität von Arzneimitteln & Gesundheitswesen (EDQM), eine Direktion des Europarats, bietet dafür wissenschaftliche und administrative Unterstützung. Das leitende Gremium des Europäischen Arzneibuchs ist das Europäische Arzneibuchkomitee, welches die Standards entwickelt und festlegt.
EU-GMP – Annex (Anhang)
Im Kontext des EU-GMP-Leitfadens stehen die Anhänge, oder "Annexes", für ergänzende Dokumente, die spezifische Vorschriften und Leitlinien für bestimmte Bereiche der Good Manufacturing Practice (GMP) bieten. Während der Hauptteil des GMP-Leitfadens allgemeine Anforderungen an die Produktionsprozesse und Qualitätskontrollen in der pharmazeutischen Herstellung abdeckt, vertiefen die Annexes bestimmte Themen oder spezialisierte Herstellungsformen.
Jeder Annex konzentriert sich auf ein anderes spezielles Thema, wie beispielsweise die Herstellung von Sterilprodukten, die Verfahrensweise bei bestimmten Technologieanwendungen oder die Untersuchung von spezifischen Qualitätsaspekten. Diese Anhänge helfen Herstellern, die besonderen Herausforderungen und Anforderungen in verschiedenen Bereichen besser zu verstehen und sicherzustellen. Aktuell stehen Annexe von Annex 1 bis Annex 20 zur Verfügung.
EU-GMP Annex 11 – Computergestützte Systeme
Der Anhang wurde überarbeitet, um dem verstärkten Einsatz und der gestiegenen Komplexität computergestützter Systeme gerecht zu werden, die in GMP-pflichtigen Vorgängen eingesetzt werden. Solche Systeme bestehen aus Software- und Hardwarekomponenten, die gemeinsam bestimmte Funktionen erfüllen. Sie müssen validiert und die IT-Infrastruktur qualifiziert werden. Wenn manuelle Tätigkeiten durch computergestützte Systeme ersetzt werden, dürfen Produktqualität, Prozesskontrolle und Qualitätssicherung nicht beeinträchtigt und das Gesamtrisiko des Prozesses nicht erhöht werden.
EU-GMP Annex 15 – Qualifizierung und Validierung
Dieses Dokument bietet Anleitungen zur Auslegung der GMP-Grundsätze für Human- und Tierarzneimittel gemäß den Richtlinien 2003/94/EG und 91/412/EWG. Es beschreibt die Qualifizierung und Validierung von Einrichtungen, Ausrüstung, Betriebsmitteln und Prozessen in der Arzneimittelherstellung, und kann optional für Wirkstoffe genutzt werden, ohne zusätzliche Anforderungen zu stellen. Hersteller müssen kritische Aspekte über den gesamten Produkt- und Prozesslebenszyklus durch Qualifizierung und Validierung kontrollieren. Änderungen, die die Produktqualität beeinflussen, sollten dokumentiert und deren Auswirkungen auf den Validierungsstatus geprüft werden. Computergestützte Systeme müssen gemäß Anhang 11 validiert werden, wobei die Leitlinien ICH Q8, Q9, Q10 und Q11 zu berücksichtigen sind.
EU-GMP Annex 20 – Qualitäts-Risikomanagement
Der Anhang 20 des EG-GMP-Leitfadens, der der ICH Q9-Leitlinie entspricht, bietet Anleitungen für ein systematisches Qualitäts-Risikomanagement. Er erleichtert die Einhaltung der GMP-Anforderungen und anderer Qualitätsstandards, indem er Grundsätze und Methoden für formales Qualitäts-Risikomanagement bereitstellt.
EU-GMP Leitfaden
Die Europäischen Kommission hat in den Grundsätzen der Guten Herstellungspraxis (GMP) für Humanarzneimittel Anforderungen an die Qualitätssicherung für Produktionsabläufe und -umgebungen festgelegt, um eine Prozessüberprüfung zu gewährleisten. Detaillierte Leitlinien für die Auslegung dieser GMP-Grundsätze sind im EU-GMP-Leitfaden zu finden.
- EU-GMP-Leitfaden Teil I: Leitfaden der Guten Herstellungspraxis.
- EU-GMP-Leitfaden Teil II: Grundlegende Anforderungen für Wirkstoffe zur
Verwendung als Ausgangsstoffe.
- EU-GMP-Leitfaden Teil III: GMP-bezogene Dokumente.
- EU-GMP-Leitfaden Teil IV: GMP-Anforderungen für Arzneimittel für neuartige
Therapien.
F0-Value (F0-Wert, Letalitätswert)
Der F0-Wert ist ein Maß für die Effektivität der Dampfsterilisation, definiert als die benötigte Zeit in Minuten bei 121,1°C zur Abtötung aller Mikroorganismen unter Berücksichtigung eines festgelegten Z-Werts.
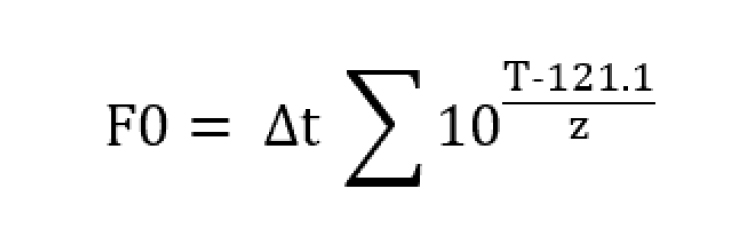
FAT – Factory-Acceptance-Test (Werksabnahme)
Die Werksabnahme, auch bekannt als Factory Acceptance Test (FAT), ist die Prüfung und Abnahme eines Produkts beim Hersteller, bevor es an den Kunden ausgeliefert wird. Ziel des FAT ist es, die Funktionalität und Qualität der Ausrüstung unter kontrollierten Bedingungen zu überprüfen, um sicherzustellen, dass sie den Spezifikationen entspricht. Nach der Lieferung an den Kunden folgt typischerweise der Site Acceptance Test (SAT), bei dem das Produkt am endgültigen Standort des Kunden getestet wird, um die ordnungsgemäße Installation und Betriebsbereitschaft zu verifizieren. Diese beiden Tests sind wichtige Schritte im Qualitätsmanagementprozess, um sicherzustellen, dass das Produkt den Anforderungen entspricht und zuverlässig funktioniert.
FDA – 482 (FDA-Inspektionsformular)
Die FDA kann einen Betrieb aus verschiedenen Gründen, wie etwa routinemäßigen Untersuchungen, Umfragen oder als Reaktion auf gemeldete Probleme, inspizieren. Bei der Ankunft im Werk wird der Ermittler seine Ausweise und das Inspektionsformular (FDA-Formular 482) vorlegen.
https://www.fda.gov/industry/fda-basics-industry/what-should-i-expect-during-inspection
FDA – 483 (FDA-Mängelbericht)
Das Formular 483 wird genutzt, um die während einer FDA-Inspektion festgestellte Mängel zu dokumentieren. Der Inspektor übergibt es bei der Abschlussbesprechung der Inspektion. Nach Übergabe wird das Formular auch an das zuständige Bezirksbüro gesendet, das für die Beurteilung der Inspektion zuständig ist. Je nach Relevanz der festgestellten Mängel kann in Folge ein Warning Letter als nächste behördliche Maßnahme folgen.
Erstellte FDA 483 Dokumente werden ebenfalls öffentlich zugänglich auf der FDA-Home Page publiziert.
https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/inspection-references/inspection-observations
FDA – Food and Drug Administration
Die FDA (Food and Drug Administration) ist eine Behörde des US-amerikanischen Gesundheitsministeriums, die für die Regulierung und Überwachung von Lebensmitteln, pharmazeutischen Medikamenten, medizinischen Geräten, Impfstoffen, biologischen Produkten, Kosmetika und Tabakerzeugnissen verantwortlich ist. Sie gewährleistet, dass diese Produkte sicher, wirksam und ordnungsgemäß gekennzeichnet sind. Darüber hinaus ist die FDA für die Zulassung und Kontrolle dieser Produkte zuständig auch für importierte Waren, die in ihren Zuständigkeitsbereich fallen.
FDA – Warning Letter (FDA-Abmahnung)
Ein FDA Warning Letter ist ein formeller Brief der U.S. Food and Drug Administration (FDA), der an Unternehmen verschickt wird, wenn diese gegen gesetzliche oder regulatorische Anforderungen verstoßen haben. Der Brief weist auf spezifische Verstöße hin, die bei einer Inspektion oder Überprüfung festgestellt wurden, und fordert das betroffene Unternehmen dazu auf, die Probleme umgehend zu beheben. Der Erhalt eines solchen Schreibens ist ernst, da eine Nichteinhaltung der gesetzten Frist zur Beseitigung der festgestellten Mängel die Ablehnung einer Zulassung oder ein vollständiger Importstopp zur Folge hat. Warning Letter werden allgemein zugänglich auf der Internetseite der FDA publiziert.
https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/compliance-actions-and-activities/warning-letters
FDA Guidance for Industry – Process Validation
Diese Richtlinie fasst die von der FDA empfohlenen Prinzipien und Ansätze zur Prozessvalidierung bei der Herstellung von Arzneimitteln und biologischen Produkten für Menschen und Tiere zusammen. Sie integriert bewährte Verfahren, die Hersteller zur Validierung ihrer Produktionsprozesse einsetzen können, und stimmt diese mit dem Konzept des Produktlebenszyklus sowie bestehenden FDA- und ICH-Richtlinien ab.
Fh-Value (FH -Wert, Wärmepenetrationsfaktor)
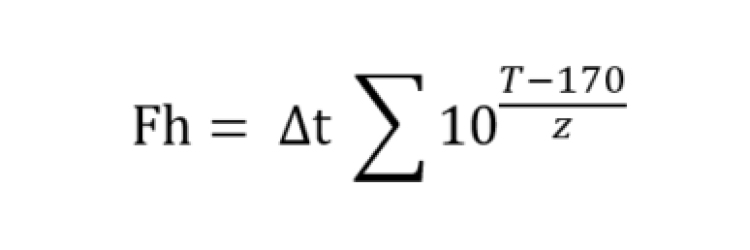
Der Fh-Wert, häufig als Wärmepenetrationsfaktor im Kontext der Sterilisation mit trockener Hitze, ist ein wichtiger Parameter zur Beurteilung der Effizienz und Wirksamkeit des Sterilisationsprozesses. Anders als bei der Sterilisation mit feuchter Hitze, bei der Dampf bei typischerweise 121,1°C verwendet wird, kommen bei den Sterilisationsverfahren mit trockener Hitze höhere Temperaturen zum Einsatz. Der Fh-Wert quantifiziert die Zeit und Temperatur, die notwendig sind, um ein bestimmtes Maß an mikrobieller Reduktion unter diesen Bedingungen zu erreichen.
FMEA – Failure Mode and Effects Analysis (Fehlermöglichkeits- und Einflussanalyse)
FMEA steht für Failure Mode and Effects Analysis. Es ist eine systematische Methode zur Identifizierung und Bewertung potenzieller Fehler in einem Produkt oder Prozess sowie deren Auswirkungen, bevor sie auftreten. FMEA hilft Risiken zu identifizieren, um proaktive Maßnahmen zur Risikominderung zu ergreifen. Die Analyse bewertet die Schwere der möglichen Fehler, die Wahrscheinlichkeit ihres Auftretens und die Erkennbarkeit der Fehler. Diese Methodik ist weit verbreitet im Qualitätsmanagement, insbesondere in Bereichen, die hohen Sicherheits- und Zuverlässigkeitsanforderungen entsprechen müssen.
FMECA – Failure Mode, Effects, and Criticality Analysis
Ist ein methodisches Verfahren der FMEA (Failure Mode and Effects Analysis), das sich nicht nur auf die Identifizierung von Fehlermodi und deren Auswirkungen konzentriert, sondern auch die Kritikalität dieser Fehlermodi bewertet.
FTA – Fault Tree Analysis (Fehlerbaum-Analyse)
Es ist eine methodische Technik zur Identifizierung und Analyse der verschiedenen Faktoren, die zu einem bestimmten Fehlerereignis im System führen können.
GAMP® – Good Automated Manufacturing Practice (Gute Automatisierte Herstellungspraxis)
Der GAMP®-Leitfaden, erstmals 1994 vom Pharmaceutical Industry Computer Systems Validation Forum (heute GAMP®-Forum) in Zusammenarbeit mit der ISPE® veröffentlicht, hat sich als Standard für die Validierung computergestützter Systeme in der pharmazeutischen Industrie etabliert. Obwohl er keine gesetzliche Bindung hat und alternative Validierungsansätze möglich sind, dient er als wichtige Orientierungshilfe für Hersteller und Zulieferer.
GAMP® 5V-Modell (V-Modell)
GAMP® 5 ist ein Leitfaden zur Validierung automatisierter Systeme in der Pharmaindustrie. Das V-Modell stellt den Entwicklungs- und Validierungsprozess in einer V-Form dar, wobei jede Entwicklungsphase auf der linken Seite einer entsprechenden Testphase auf der rechten Seite gegenübersteht. Das Modell hilft, sicherzustellen, dass Systeme umfassend überprüft und regulative Standards eingehalten werden.
Durch die sequenzielle Vorgehensweise wird sichergestellt, dass alle Dokumente und Validierungsschritte in einer logischen Folge abgearbeitet werden. Zuständigkeiten und Verantwortlichkeiten zwischen Zulieferer und Auftraggeber können ebenfalls im V-Modell definiert werden. Somit werden neben den technischen auch organisatorische Aspekten erfasst und erleichtert die Zusammenarbeit der an einem Projekt beteiligten Parteien.

GCP – Good Clinical Practice
Umfasst international anerkannte Richtlinien, die ethische und wissenschaftliche Standards für die Planung und Durchführung klinischer Studien festlegen.
GDP – Good Distribution Practice
Beinhaltet Maßnahmen und Richtlinien, die für den ordnungsgemäßen Vertrieb von pharmazeutischen Produkten wichtig sind. Diese Praktiken sind darauf ausgelegt, die Qualität und Integrität von Arzneimitteln zu bewahren, während sie durch die Lieferkette transportiert werden. GDP deckt Aspekte wie Lagerung, Transport, Rückverfolgbarkeit und Management von Qualitätsrisiken ab. Ziel ist es sicherzustellen, dass die Produkte sicher, effektiv und unversehrt bei den Endverbrauchern ankommen.
GEP – Good Engineering Practice
Umfasst eine Reihe von Standards und Verfahren, die bei der Planung, Konstruktion, Implementierung und Wartung technischer Systeme und Projekte angewendet werden. Diese Praktiken zielen darauf ab, sicherzustellen, dass alle technischen Lösungen und Anlagen effizient, sicher und konform mit regulatorischen Anforderungen betrieben werden.
GLP – Good Laboratory Practice
Gute Laborpraxis (GLP) ist ein Regelwerk, das Anforderungen an die Organisation, Planung und Durchführung von nicht-klinischen Studien mit Substanzen und Arzneimitteln stellt. Es umfasst auch die Dokumentation der Ergebnisse sowie die Qualitätskontrolle dieser Prüfungen.
GMP – Good Manufacturing Practice (Gute Herstellungspraxis) / cGMP – current Good Manufacturing Practice (Aktuelle Gute Herstellungspraxis)
Beschreibt generell Vorgaben und Richtlinien zur Sicherstellung einer kontinuierlich hohen Produktqualität von Arzneimitteln und Wirkstoffen.
GSP – Good Storage Practices
Gute Lagerungspraxis (GSP) umfasst Standards und Richtlinien, die darauf abzielen, Produkte sicher, effizient und qualitätserhaltend zu lagern.
GxP/ cGxP
GxP steht für "Good x Practice" und ist ein allgemeiner Begriff, der sich auf verschiedene Qualitätsrichtlinien und -vorschriften bezieht, die in regulierten Industrien wie der Pharma-, Lebensmittel- und Medizinproduktebranche angewendet werden. Das "x" in GxP kann durch verschiedene Buchstaben ersetzt werden, um unterschiedliche Bereiche abzudecken:
- GLP: Good Laboratory Practice (Gute Laborpraxis) – bezogen auf die Qualität und Integrität von Labordatenergebnissen.
- GMP: Good Manufacturing Practice (Gute Herstellungspraxis) – bezogen auf die Qualitätssicherung während der Produktion.
- GDP: Good Distribution Practice (Gute Vertriebspraxis) – bezieht sich auf die ordnungsgemäße Lagerung und Transport von Produkten.
- GCP: Good Clinical Practice (Gute klinische Praxis) – bezogen auf die ethischen und wissenschaftlichen Standards für klinische Studien.
- GEP: Good Engineering Practice (Gute Ingenieurpraxis) – bezieht sich auf die bewährten Vorgehensweisen bei technischen Projekten.
HACCP – Hazard Analysis and Critical Control Points (Gefahrenanalyse und kritische Kontrollpunkte)
HACCP ist ein proaktives Risikomanagementsystem, das entwickelt wurde, um die Sicherheit von Lebensmitteln und pharmazeutischen Produkten zu gewährleisten. Das HACCP-System identifiziert potenzielle Gefahren in einem Produktionsprozess und legt kritische Kontrollpunkte fest, an denen Maßnahmen eingeleitet werden können, um diese Gefahren zu minimieren oder zu eliminieren.
HAZOP – Hazard and Operability Study (Gefahren- und Funktionsfähigkeitsstudie)
Diese Methode ist ein systematischer Ansatz zur Identifizierung potenzieller Risiken und Schwachstellen in Industrieprozessen, vor allem in der chemischen und pharmazeutischen Industrie. HAZOP-Studien werden durchgeführt, um Abweichungen von der geplanten Betriebsweise zu erkennen, die zu Gefahren führen könnten, und um die Auswirkungen auf die Sicherheit, Effizienz und Qualität zu bewerten. Das Verfahren beinhaltet die strukturierte Analyse von Prozessen anhand von "Guide Words", um mögliche Gefahrenpunkte und fehlerhafte Betriebsbedingungen zu identifizieren.
ICH – International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use
Die ICH, ursprünglich die "International Conference on Harmonization", wurde 1990 von der FDA, der Europäischen Kommission, dem japanischen Gesundheitsministerium sowie großen Arzneimittel-Herstellerverbänden gegründet. Mit Sitz in Genf erstellt die ICH standardisierte Guidelines zur Qualität, Wirksamkeit und Sicherheit von Arzneimitteln, wie GCP oder GMP. Diese Guidelines dienen als Leitlinien für die Pharmaindustrie. Beobachter sind die WHO (World Health Organization / Weltgesundheitsorganisation) oder EFTA (European Free Trade Association/ Europäische Freihandelsassoziation). In der EU werden die Richtlinien vom EMA (European Medicines Agency/ Europäische Arzneimittel-Agentur) übernommen und gelten als Standards, von denen Pharmaunternehmen nur in Ausnahmefällen abweichen sollten.
ICH Q9 – Quality Risk Management – Scientific Guideline
Dieses Dokument bietet Prinzipien und Beispiele für das Qualitätsrisikomanagement, die auf verschiedene Aspekte von Qualitätssicherungsmaßnahmen angewendet werden können. Dazu gehören die Entwicklung, Herstellung, Verteilung sowie Inspektions- und Einreichungs-/Überprüfungsprozesse während des gesamten Lebenszyklus von Wirkstoffen, Arzneimitteln, biologischen und biotechnologischen Produkten.
Zusammengefasst: Das Dokument enthält Richtlinien und Werkzeuge für das Qualitätsrisikomanagement, die in der gesamten Wertschöpfungskette pharmazeutischer Produkte anwendbar sind.
IQ – Installation Qualification (Installationsqualifizierung)
Diese Phase des Validierungsprozesses wird durchgeführt, um sicherzustellen, dass Ausrüstungen, Systeme oder Anlagen korrekt installiert sind und alle technischen Spezifikationen, Herstelleranforderungen sowie regulatorischen Standards erfüllen. Dazu gehören u.a. die Abnahme einer fachgerechten Elektroinstallation, die detaillierte Überprüfung aller Ein- und Ausgangssignale sowie die korrekte Berechnung ermittelter Messwerte.
ISPE® – International Society for Pharmaceutical Engineering
Die International Society for Pharmaceutical Engineering ist eine gemeinnützige Organisation, die wissenschaftliche, technische und regulatorische Fortschritte in der Pharmaindustrie fördert. Sie unterstützt ihre Mitglieder durch Wissensaustausch, Schulungen und Ressourcen zu ingenieurtechnischen und reglementierten Prozessen und ist an der Ausarbeitung von Normen und Regularien beteiligt.
ISPE® GAMP® 5 – Risikobasierter Ansatz für konforme computergestützte Systeme
GAMP® 5 (Good Automated Manufacturing Practice) ist ein Leitfaden, der bei der Implementierung und Validierung computergestützter Systeme in der pharmazeutischen Industrie verwendet wird. Er bietet einen risikobasierten Ansatz, um sicherzustellen, dass alle automatisierten Systeme in Übereinstimmung mit den geltenden regulatorischen Anforderungen betrieben werden und die beabsichtigte Produktqualität und Datensicherheit gewährleisten. GAMP® 5 betont die Bedeutung eines durchgängigen Lebenszyklusansatzes für die Validierung von Systemen, wobei die Projektplanung, Spezifikation, Verifizierung und Wartung berücksichtigt werden.
JP – Japanese Pharmacopeia (Arzneibuch Japan)
Das japanisches Arzneibuch (Japanese Pharmacopoeia, JP) stellt Standards für die Qualität, Sicherheit und Wirksamkeit von Arzneimitteln in Japan bereit. Es enthält verbindliche Normen für die Herstellung, Prüfung und Verwendung von pharmazeutischen Produkten.
LVP – Large Volume Parenteral
LVPs sind sterile Arzneimittel, die in Behältern mit mehr als 100 ml abgefüllt sind. Diese Produkte werden hauptsächlich zur intravenösen Verabreichung von Flüssigkeiten mit Elektrolyten, Nährstoffen oder Medikamenten verwendet. LVPs durchlaufen strenge Sterilisationsverfahren, oder sogenannte aseptische Filtration, um ihre Sicherheit und Wirksamkeit sicherzustellen.
OOE – Out-of-Expectation (Außerhalb der Erwartungen)
„Out of Expectation“ (OOE) Ergebnisse sind Befunde, die sich als einmalige Anomalien erweisen und statistisch gesehen keine signifikante Bedeutung haben. Sie deuten in der Regel nicht auf ein systemisches Problem hin. OOE-Ergebnisse sind typischerweise untypische oder abweichende Befunde, die nicht mit anderen vorhandenen Daten übereinstimmen. Im Gegensatz zu einem „Out of Specification“ (OOS) Ergebnis verletzen OOE-Befunde keine festgelegten Spezifikationsgrenzen.
OOS – Out-of-Specification (Außerhalb spezifizierter Akzeptanzkriterien)
Out of Specification (OOS) bezieht sich auf Testergebnisse, die außerhalb der festgelegten Akzeptanzkriterien liegen, wie sie in offiziellen Arzneibüchern oder firmeneigenen Dokumenten definiert sind. Der Begriff wird vor allem im Zusammenhang mit den Regularien der FDA verwendet. OOS-Ergebnisse sind Testergebnisse, die nicht den spezifizierten Standards entsprechen. Die Behandlung solcher Abweichungen ist ein wesentlicher Bestandteil von Qualitätssicherungssystemen wie ISO 9001, GMP und GLP.
OOT – Out-of-Trend
OOT beschreibt ein Ergebnis, das innerhalb der vorgegebenen Grenzwerte liegt, jedoch einen Trend zeigt, der in der statistischen Auswertung auffällig ist. Solche Ergebnisse werden oft als "Out of Trend" (OOT) bezeichnet. Ein OOT-Ergebnis weist darauf hin, dass während die Ergebnisse noch innerhalb der festgelegten Spezifikationen liegen, es dennoch eine Tendenz oder Veränderung über die Zeit gibt, die Aufmerksamkeit erfordert. Solche Trends können wichtig sein, um potenzielle Probleme frühzeitig zu erkennen und sicherzustellen, dass die Prozesse stabil bleiben.
OQ – Operational Qualification (Funktionsqualifizierung)
Die OQ, oder Funktionsqualifizierung (Operational Qualification), ist ein kritischer Schritt im Validierungsprozess von Anlagen und Systemen innerhalb regulierter Industrien, wie etwa der Pharma- und Medizintechnikbranche. Sie überprüft und dokumentiert, dass alle Ausrüstungen, Systeme oder Prozesse in ihrer vorgesehenen Umgebung über ihren gesamten Betriebsbereich hinweg ordnungsgemäß funktionieren. Während der OQ-Phase werden spezifische Testprotokolle und Verfahren durchgeführt, um sicherzustellen, dass alle Betriebsparameter den festgelegten Spezifikationen entsprechen.
PAT – Process Analytical Technology (Prozessanalysentechnologie)
In der pharmazeutischen und biotechnologischen Industrie bezieht sich die Prozessanalysentechnologie (PAT) auf ein System zur Analyse und Kontrolle der Fertigungsprozesse durch Messungen von kritischen Qualitätsmerkmalen (CQAs) des Produkts in Echtzeit. Ziel der PAT ist es, ein besseres Verständnis und eine bessere Kontrolle der Herstellungsprozesse zu erlangen, um die Produktqualität zu optimieren und gleichzeitig die Produktionskosten und -zeiten zu senken.
PCS – Process Control System (Prozesssteuerungssystem)
Ein Prozesssteuerungssystem ist ein integriertes System von Technologien und Verfahren, das zur Überwachung und Steuerung von Produktionsprozessen eingesetzt wird. Es gewährleistet, dass diese Prozesse effizient, sicher, und innerhalb der spezifizierten Parameter ablaufen. In der pharmazeutischen Herstellung wird ein PCS genutzt, um sicherzustellen, dass sowohl Zwischenprodukte als auch fertige Produkte, wie zum Beispiel Wirkstoffe (APIs), Spezifikationen und Qualitätsstandards entsprechen.
PDA® Parenteral Drug Association
Die Parenteral Drug Association (PDA®) ist eine internationale Organisation, die sich auf die Förderung von Wissenschaft und Regulierung in der pharmazeutischen und biotechnologischen Industrie konzentriert. Sie unterstützt Fachleute in diesen Bereichen, indem sie Bildung, Fachwissen und Richtlinien bereitstellt, um die Qualität und Sicherheit von parenteralen (d.h. nicht-oral verabreichten) Medikamenten und verwandten Produkten zu verbessern. Die PDA® ist bekannt für ihre Rolle bei der Entwicklung von Best Practices und Standards. Die sogenannten Technical Reports bieten praktische Anleitungen für verschiedenste Anwendungen und werden unter anderem in Normen und Richtlinien referenziert.
PHA – Preliminary Hazard Analysis (Vorgezogene Gefahrenanalyse)
Diese Methode ist eine frühe Form der Risikoanalyse, die in der Planungsphase eines Projekts oder bei der Einführung neuer Prozesse und Produkte eingesetzt wird. Ziel der PHA ist es, potenzielle Gefahren zu identifizieren und Risiken abzuschätzen, bevor das Design finalisiert wird. Die Analyse betrachtet mögliche Ereignisse, die zu Gefahren führen könnten, sowie deren Auswirkungen und mögliche Maßnahmen zur Risikominderung.
PIC/S® – Pharmaceutical Inspection Cooperation Scheme
Das Pharmaceutical Inspection Co-operation Scheme (PIC/S®) ist ein internationales Kooperationsprogramm, das ursprünglich als Nachfolgeprogramm der Pharmaceutical Inspection Convention ins Leben gerufen wurde. Ziel von PIC/S® ist es, die Zusammenarbeit und Harmonisierung zwischen den Good Manufacturing Practice (GMP)-Überwachungsbehörden weltweit zu fördern. Dies geschieht, indem die Inspektionsprozesse vereinheitlicht und die Kommunikation zwischen den Regulierungsbehörden und der pharmazeutischen Industrie verbessert werden. Durch die Veröffentlichung von Leitlinien und Empfehlungen trägt PIC/S® dazu bei, die Herstellung und Sicherheit pharmazeutischer Produkte auf globaler Ebene sicherzustellen.
Die umfassenden Standards, die PIC/S ®bereitstellt, helfen dabei, die Qualitätssicherung zu verbessern und sogenannte Mehrfachinspektionen zu vermeiden.
PPQ – Process Performance Qualification (Prozessleistungsqualifizierung)
Die Prozessleistungsqualifizierung (PPQ) ist ein zentraler Schritt in der Prozessqualifizierung, der zweiten Phase der Prozessvalidierung. Sie geht über die Performance Qualification hinaus, indem sie die langfristige Leistung und Robustheit des Herstellungsprozesses bewertet. Laut GAMP® 5 erweitert die PPQ die Qualifizierung einer Anlage zur Produktion von Medizin- und Pharmaprodukten nach einer abgeschlossenen Performance Qualification.
PQ – Performance Qualification (Leistungsqualifizierung)
Die Leistungsqualifizierung (Performance Qualification, PQ) ist der dokumentierte Nachweis, dass Anlagen, Systeme und Ausrüstungen effizient und mit wiederholbarer Leistung gemäß den genehmigten Anforderungen arbeiten. Dazu gehören Tests unter realen Bedingungen, die Bestätigung der Einhaltung von Qualitätsstandards, die Bewertung der Reproduzierbarkeit von Ergebnissen sowie die sorgfältige Datendokumentation und -analyse.
PQR – Product Quality Review (Produktqualitätsüberprüfung)
Die Produktqualitätsüberprüfung (PQR) ist eine gesetzlich vorgeschriebene, periodische Bewertung, die sicherstellt, dass pharmazeutische Produkte gemäß cGMP-Anforderungen konstant nach Qualitäts-, Sicherheits- und Wirksamkeitsstandards hergestellt und kontrolliert werden. Neben der retrospektiven Analyse dient der PQR auch der Echtzeit-Prozesskontrolle und kontinuierlichen Verbesserung, indem er eine umfassende Sicht auf alle relevanten Daten bietet. Durch die Anwendung statistischer Werkzeuge werden Abweichungen vorhergesagt und Trends erkannt, sodass bei Bedarf korrektive und präventive Maßnahmen ergriffen werden können. Die Frequenz der Durchführung basiert u.a. auf historischen Überprüfungsergebnissen sollte aber mindestens einmal jährlich durchgeführt werden.
Predictive Maintenance (Prädiktive Wartung)
Diese Methode nutzt fortschrittliche Technologien zur Echtzeitüberwachung und Datenanalyse, um vorherzusagen, wann Wartung erforderlich ist. Dies ermöglicht das Erkennen potenzieller Ausfälle und die Durchführung von Wartungsarbeiten nur bei Bedarf, was die Gerätelebensdauer verlängert und Ausfallzeiten sowie Wartungskosten reduziert.
Als Beispiel hierfür dienen kontinuierliche Monitoringsysteme wie z.B. das Kaye LabWatch System.
Preventive Maintenance (Präventive Wartung)
Präventive Wartung beinhaltet planmäßige Wartungsarbeiten, die unabhängig vom Gerätezustand durchgeführt werden, um Ausfälle zu verhindern und die Lebensdauer der Anlagen zu verlängern.
Prospective Validation or Premarket Validation (Prospektive Validierung)
Die prospektive Validierung umfasst Validierungsaktivitäten, die durchgeführt werden, bevor ein Produkt routinemäßig hergestellt und zum Verkauf freigegeben wird. Ziel ist es, sicherzustellen, dass ein Prozess oder System vor der kommerziellen Nutzung die erforderlichen Qualitäts-, Sicherheits- und Wirksamkeitsstandards erfüllt. Dieser Prozess beinhaltet die Planung, Durchführung und Dokumentation von Tests zur Einhaltung regulatorischer Anforderungen.
PS – Purified Steam (Reindampf)
Reindampf ist Dampf, der die strengen Reinheitskriterien erfüllt, um als Wasser für Injektionszwecke (Water For Injection, WFI) zu gelten, sobald er kondensiert ist. In der pharmazeutischen Industrie wird Reindampf vor allem zur Sterilisation von Bauteilen verwendet, die mit Produkten in Berührung kommen. Außerdem dient er zur Befeuchtung der Luftzufuhr in Reinräumen und Isolatoren, um eine kontrollierte und sterile Umgebung zu gewährleisten.
PV – Process Validation (Prozessvalidierung)
Die Prozessvalidierung ist ein schrittweises und dokumentiertes Verfahren, um sicherzustellen, dass Prozesse innerhalb ihrer festgelegten Designparameter in der Lage sind, ein Endprodukt von der erforderlichen Qualität konsistent zu produzieren. Der Umfang der Validierung wird maßgeblich durch eine zuvor durchgeführten Risikoanalyse bestimmt. Je größer das Risiko, desto höher der Validierungsaufwand.
PW – Purified Water (Gereinigtes Wasser)
Gereinigtes Wasser (Purified Water) ist Wasser, das durch verschiedene Verfahren, wie Destillation, Umkehrosmose oder Ionenaustausch, von chemischen und mikrobiologischen Verunreinigungen befreit wurde. Es wird vor allem in der pharmazeutischen Industrie eingesetzt, wo hohe Reinheitsanforderungen bestehen, jedoch keine Sterilität oder Pyrogenfreiheit notwendig sind. Gereinigtes Wasser wird in der Herstellung nicht-injizierbarer Arzneimittel, als Lösungsmittel oder als Basis für die Herstellung von Produkten wie Dialyselösungen verwendet, vorausgesetzt, es erfüllt bestimmte Reinheitsanforderungen, wie etwa den Endotoxintest gemäß den pharmakopöischen Standards.
QA – Quality Assurance (Qualitätssicherung QS)
Qualitätssicherung (QA) ist ein systematischer Prozess (Prozess Fokus) zur Überwachung und Verbesserung von Qualitätsstandards in der Herstellung und Bereitstellung von Produkten oder Dienstleistungen. In der pharmazeutischen Industrie stellt QA sicher, dass alle Produkte die erforderlichen Qualitätsstandards und regulatorischen Anforderungen erfüllen, angefangen bei der Überprüfung der Rohstoffqualität bis hin zur umfassenden Prüfung der Endprodukte.
QbD – Quality-by- Design (Qualität durch Design)
Die Europäische Arzneimittel-Agentur (EMA) unterstützt die Anwendung von Qualität durch Design (Quality by Design), um die Arzneimittelherstellung zu optimieren. Dieser Ansatz sichert die Qualität von Arzneimitteln durch statistische und analytische Methoden sowie Risikomanagement. Dabei werden Variabilitätsquellen identifiziert und gesteuert, um sicherzustellen, dass Medikamente von Anfang an den vordefinierten Eigenschaften entsprechen. Das Konzept nutzt multivariate Analysen und moderne Tools, um kritische Merkmale und Produktionsparameter besser zu verstehen und kontinuierliche Verbesserungen zu ermöglichen.
QC – Quality Control (Qualitätskontrolle)
Qualitätskontrolle (QC) ist ein systematischer Prozess in der Fertigungsindustrie, der sicherstellt, dass Produkte (Produkt Fokus) die festgelegten Qualitätsstandards erfüllen. Die QC umfasst die Prüfung und Inspektion von Produkten in verschiedenen Phasen der Produktion, um sicherzustellen, dass sie den Spezifikationen entsprechen und frei von Defekten sind. Dieser Prozess beinhaltet die Identifikation und Korrektur von Mängeln sowie die Überprüfung der Einhaltung von Vorschriften und Standards.
QMS – Quality Management System (Qualitätsmanagementsystem)
Ein Qualitätsmanagementsystem (QMS) im Bereich der Guten Herstellungspraxis (GMP) ist ein strukturiertes System von Verfahren, Prozessen und Ressourcen, die darauf ausgelegt sind, die Qualität und Sicherheit von pharmazeutischen Produkten während des gesamten Produktionsprozesses sicherzustellen. Es umfasst alle Elemente des Qualitätsmanagements, die Planung, Kontrolle, Qualitätssicherung und kontinuierliche Verbesserung zur Einhaltung von regulatorischen Anforderungen und zur Gewährleistung der Wirksamkeit und Sicherheit von Arzneimitteln.
QP – Qualification Protocol (Qualifizierungsprotokoll)
Ein Qualifizierungsprotokoll (QP) ist ein schriftlicher Plan oder ein Verfahren, das im Detail beschreibt, wie die Qualifizierung erreicht wird. Es umfasst spezifische Qualifizierungsanforderungen für jedes Gerät, jede Systemanforderung und jede Produktanforderung.
QP – Qualified Person (Sachkundige Person)
Die Sachkundige Person in der Pharmazie ist eine Schlüsselrolle im europäischen Arzneimittelrecht, verantwortlich für die Herstellung, Prüfung und Freigabe von Arzneimitteln gemäß den Vorschriften. Sie sorgt zudem für die vollständige Dokumentation der Einhaltung gesetzlicher Anforderungen.
QSM – Quality System Manual (Qualitätssicherungshandbuch QSH)
Ein Qualitätssicherungshandbuch ist ein umfassendes Dokument, das die Struktur, Prozesse und Richtlinien des Qualitätsmanagementsystems (QMS) eines Unternehmens beschreibt. Es legt die Standards und Verfahren fest, die sicherstellen sollen, dass Produkte und Dienstleistungen die erforderlichen Qualitätsanforderungen erfüllen. Das Handbuch dient als Leitfaden für alle Mitarbeiter, um die Qualitätsziele des Unternehmens zu erreichen und die Einhaltung von rechtlichen und branchenspezifischen Anforderungen zu gewährleisten.
Qualification (Qualifizierung)
Der Begriff "Qualifizierung" im GxP-Umfeld bezeichnet den Prozess der Überprüfung der Eignung eines Geräts, einer Anlage oder eines Systems für seinen vorgesehenen Zweck. Die erste Erwähnung von "Qualifizierung" in GxP-Vorschriften lässt sich schwer genau bestimmen, wird jedoch auf die späten 1970er Jahre zurückgeführt, als die FDA die Validierung als Teil der Guten Herstellungspraxis (GMP) einführte. Dieser Prozess umfasst Design-, Installations-, Funktions- und Leistungsqualifizierungen (DQ, IQ, OQ, PQ). Der genaue Qualifizierungsprozess kann jedoch je nach Kontext und spezifischem Gerät oder System variieren. Die Qualifizierung ist in einzelne, durchführbare Schritte unterteilt. Sie ist ein Bestandteil der Validierung und bezieht sich auf die Leistung und die Eigenschaften der Systeme, Geräte, Instrumente, Anlagen und Software. Üblicherweise umfasst sie folgende Schritte:
- URS (User Requirement Specification): Dokument, das die Anforderungen für ein bestimmtes System oder Produkt erfasst und definiert.
- DQ (Design Qualification): Verifizierungsprozess, der bestätigt, dass das Design der Ausrüstung oder des Systems allen vordefinierten Anforderungen für den vorgesehenen Zweck entspricht.
- FAT/SAT (Factory Acceptance Test/Site Acceptance Test): FAT ist ein Prüfprozess vor dem Versand, der am Standort des Herstellers durchgeführt wird und das Design sowie die Betriebseinhaltung des Systems bestätigt. SAT validiert die korrekte Installation und funktionale Leistung des Systems am Standort des Kunden, bevor der volle Betrieb aufgenommen wird.
- IQ (Installation Qualification): Überprüfung, dass die Ausrüstung oder das System korrekt installiert wurde und innerhalb der geplanten Parameter in der vorgesehenen Umgebung funktioniert.
- OQ (Operational Qualification): Überprüfung, dass die Ausrüstung oder das System im gesamten Betriebsbereich in ihrem spezifischen Umfeld wie beabsichtigt funktioniert.
• PQ (Performance Qualification): Bestätigung und Dokumentation, dass die Ausrüstung oder das System konsistent gemäß den im URS (User Requirement Specifications) definierten Parametern Leistungen erbringen kann.
Retrospective Validation (Retrospektive Validierung)
Die retrospektive Validierung überprüft und analysiert bestehende Produktionsdaten und Chargenaufzeichnungen, um die Konsistenz und Leistungsfähigkeit eines bereits genutzten Prozesses zu bewerten. Ziel ist es, festzustellen, ob der Prozess kontinuierlich Produkte mit den gewünschten Qualitätsmerkmalen hergestellt hat und den Qualitäts- und regulatorischen Standards entspricht, basierend auf historischen Daten.
Revalidation (Revalidierung)
Revalidierung ist der Prozess der erneuten Überprüfung und Wiederholung von Validierungsmaßnahmen, um sicherzustellen, dass Systeme, Prozesse oder Geräte weiterhin die festgelegten Qualitäts- und Leistungsstandards erfüllen. Sie umfasst die Analyse bestehender Leistungsdaten und ist entscheidend, um den validierten Status aufrechtzuerhalten, insbesondere bei signifikanten Änderungen oder um die kontinuierliche Einhaltung von regulatorischen Anforderungen zu gewährleisten.
Risk Acceptance (Risikobereitschaft)
Risikobereitschaft bedeutet, ein Risiko zu akzeptieren, entweder formell oder passiv. Auch bei guten Risikomanagementpraktiken können nicht alle Risiken beseitigt werden. Daher wird das Risiko oft auf ein spezifiziertes, akzeptables Niveau reduziert, das je nach Situation festgelegt wird.
Risk Analyse (Risikoanalyse)
Risikoanalyse ist die Einschätzung des Risikos, das mit den identifizierten Gefahren verbunden ist. Sie ist der qualitative oder quantitative Prozess der Verbindung zwischen der Eintrittswahrscheinlichkeit und der Schwere der Schäden. In einigen Risikomanagement-Tools spielt auch die Fähigkeit, den Schaden zu erkennen (Erkennbarkeit), eine Rolle bei der Einschätzung des Risikos.
Risk Assessment (Risikobewertung)
Bewertung des Eintretens eines erkannten Risikos und Bewertung ihrer Wahrscheinlichkeit und Auswirkungen. Generelle Fragestellungen:
- Was könnte schiefgehen?
- Wie wahrscheinlich ist es, dass es schiefgeht?
- Was sind die Konsequenzen (Schweregrad)?
Risk Communication (Risikokommunikation)
Austausch von Informationen über Risiken und Risikomanagement zwischen allen am Prozess beteiligten Abteilungen, der Qualitätskontrolle, dem Qualitätsmanagement und der Geschäftsleitung.
Risk Control (Risiko Kontrolle)
Festlegung von Risikoakzeptanzkriterien, Risikominderung und Risikoakzeptanz.
Die Risikokontrolle umfasst die Entscheidungsfindung zur Reduzierung und/oder Akzeptanz von Risiken. Ziel der Risikokontrolle ist es, das Risiko auf ein akzeptables Niveau zu senken. Der Aufwand für die Risikokontrolle sollte proportional zur Bedeutung des Risikos sein.
Die Risikokontrolle könnte sich auf folgende Fragen konzentrieren:
- Liegt das Risiko über einem akzeptablen Niveau?
- Was kann getan werden, um Risiken zu reduzieren oder zu beseitigen?
- Was ist das angemessene Gleichgewicht zwischen Nutzen, Risiken und Ressourcen?
- Werden neue Risiken eingeführt, da die identifizierten Risiken kontrolliert werden?
Risk Evaluation (Risikobewertung)
Risikobewertung vergleicht die identifizierten und analysierten Risiken mit festgelegten Risikokriterien.
Risk Identifikation (Risikoermittlung)
Identifizieren. erfassen und dokumentieren potenzieller Risiken, die die Qualität und Sicherheit beeinflussen könnten.
Risk Management (Risikomanagement)
Risikomanagement ist ein zentraler Bestandteil im GxP-Umfeld über den gesamten Lebenszyklus einer Anlage/ Prozess/ Systems/ Instrument und ist Bestandteil von Richtlinien und Vorschriften. Der Risikomanagementprozess soll sicherstellen, dass alle Risiken identifiziert, bewertet und angemessen kontrolliert werden, um die Qualität und Sicherheit von pharmazeutischen Produkten zu gewährleisten. Die fünf grundlegenden Schritte des Risikomanagements sind:
- Risk Identifikation (Risikoermittlung)
- Risk Assessment (Risikobewertung)
- Risk Control (Risiko Kontrolle)
- Risk Communication (Risikokommunikation)
- Risk Review (Risikokontrolle)
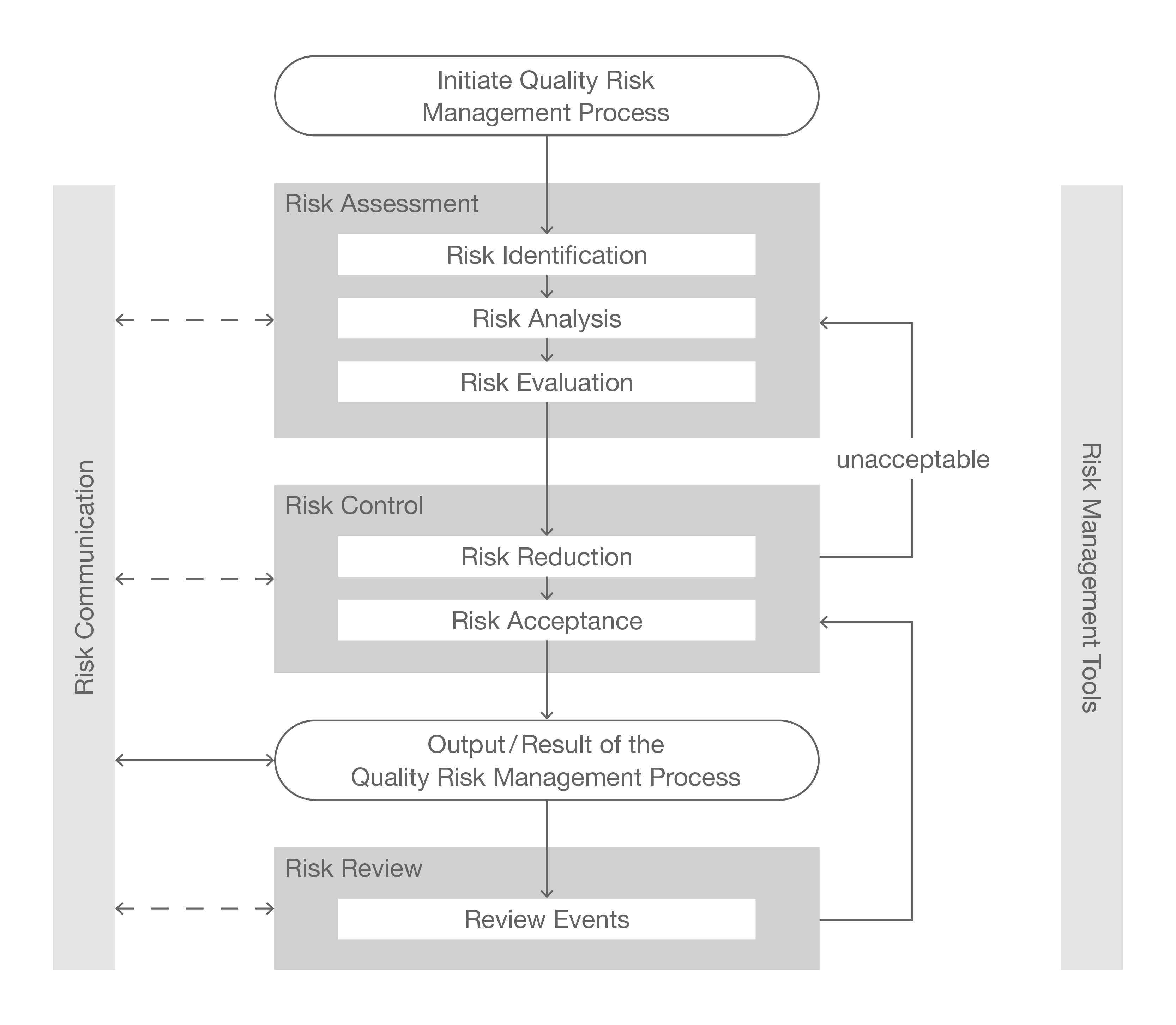
Quelle: EMA ICH guideline Q9 Quality Risk Management
Risk Management Tools (Risikomanagementmethodik)
Qualitätsrisikomanagement bietet wissenschaftliche und praktische Methoden zur Entscheidungsfindung im Risikomanagement, basierend auf der Bewertung von Wahrscheinlichkeit, Schwere und Erkennbarkeit von Risiken. Traditionelle und moderne Risikomanagement-Tools können kombiniert angewendet werden, um Flexibilität zu bieten. Diese Methoden werden an die Komplexität und Kritikalität der zu behandelnden Themen angepasst. Eine Reihe von Tools wie FMEA, FTA, HACCP, PHA und HAZOP unterstützt diesen Prozess.
Risk Reduction (Risikoreduzierung)
Risikoreduzierung beinhaltet Maßnahmen zur Verringerung oder Vermeidung von Qualitätsrisiken, wenn diese über ein akzeptables Niveau hinausgehen. Während Maßnahmen dazu beitragen, die Schwere und Wahrscheinlichkeit von Schäden zu senken und die Erkennbarkeit zu verbessern, können sie auch neue Risiken einführen oder bestehende Risiken beeinflussen. Daher sollte die Risikobewertung nach der Umsetzung von Risikominderungsmaßnahmen überprüft werden.
Risk Review (Risikoüberprüfung)
Bezieht sich auf die regelmäßige Überprüfung und Bewertung des Risikomanagementprozesses und seiner Ergebnisse, um sicherzustellen, dass dieser aktuell und effektiv bleibt. Risikomanagement sollte kontinuierlich in den Qualitätsmanagementprozess integriert sein, mit Mechanismen zur Überwachung und kontinuierlichen Bewertung. Die Ergebnisse sollten regelmäßig überprüft und an neues Wissen angepasst werden. Änderungen, wie geplante Inspektionen oder ungeplante Rückrufe, sollten in den Prozess einfließen, basierend auf den Ergebnissen der Risikoüberprüfung.
SAL – Sterility Assurance Level (Sterilitätssicherheitsniveau)
Das Sterilitätssicherheitsniveau, auch als Sterilitätssicherheitswert bekannt (englisch: Sterility Assurance Level, SAL oder SAL-Wert), beschreibt die Wahrscheinlichkeit, dass ein einzelnes Produkt nach dem Sterilisierungsprozess weiterhin kontaminiert ist. Diese Messgröße wird vor allem in der Herstellung von sterilen Arzneimitteln und Medizinprodukten verwendet.
Sanitization (Sanitisierung)
Die Sanitisierung ist ein mehrstufiger Prozess und kann üblicherweise in mehrere, typische Stufen unterteilt werden:
- Vorbereitung: Entfernen von unnötigem Material und Bereitstellung von Reinigungs- und Desinfektionsmitteln.
- Reinigung: Gründliches Entfernen von Schmutz und organischen Materialien von Oberflächen.
- Spülen: Entfernung von Reinigungsmittelrückständen durch gründliches Spülen.
- Desinfektion: Auftragen von Desinfektionsmitteln zur Abtötung verbliebener Mikroorganismen; bei Kalt-Sanitisierung werden chemische Mittel bei niedrigen Temperaturen eingesetzt.
- Kontrolle und Dokumentation: Überprüfung der Sanitisierung und Dokumentation aller Maßnahmen zur Sicherstellung der Hygienestandards.
Die Unterschiede zwischen Heiß- und Kalt-Sanitisierung liegen im Temperaturbereich und der Methode zur Mikroorganismenreduktion:
- Heiß-Sanitisierung nutzt Temperaturen zwischen 80-90°C, wie heißes Wasser oder Dampf, um Bakterien, Viren und Pilze abzutöten, ist jedoch energieintensiv und kann Materialverschleiß verursachen.
- Kalt-Sanitisierung verwendet chemische Mittel bei niedrigen Temperaturen, ideal für temperaturempfindliche Materialien, erfordert jedoch sorgfältige Auswahl und Handhabung der Chemikalien.
SAT – Site-Acceptance-Test (Abnahmetest vor-Ort)
Ein Site Acceptance Test (SAT) ist ein wichtiger Prüfprozess, der sicherstellt, dass eine Maschine oder Anlage am Kundenstandort ordnungsgemäß installiert, konfiguriert und funktionsfähig ist. Er bewertet, ob die Ausrüstung die Betriebsanforderungen erfüllt und dokumentiert den Ist-Zustand, um Abweichungen von Spezifikationen zu erfassen. Der Prozess beinhaltet die Überprüfung der Installation, die Funktionalität der Komponenten, die Einhaltung von Sicherheitsstandards und Leistungstests, um Qualität und Sicherheit vor dem regulären Betrieb zu gewährleisten.
SIP – Steam in Place (Sterilisierung vor Ort)
Steam-in-Place (SIP) Sterilisation ist ein kritischer Prozess in der Pharmaindustrie, um die Sterilität fest installierter Anlagen wie Tanks, Rohrleitungen und Bioreaktoren in der aseptischen Herstellung sicherzustellen. Dieser Prozess nutzt gesättigten Dampf zur Abtötung von Mikroorganismen, ohne dass eine Demontage der Anlagen erforderlich ist. Hierbei kommen die gleichen Normen und Regelwerke zur Anwendung wie bei einer Dampf- Autoklave/ Dampfsterilisator. Einschlägige Normen zur Sterilisation mit feuchter Hitze, wie z.B. EN 285 oder ISO 17665, werden berücksichtigt.
Als Vorstufe des SIP-Prozesses, also der Sterilisation der Anlage, erfolgt üblicherweise der CIP-Prozess (Clean-In-Place).
SMF – Site Master File (Standortdossier)
Ein Site Master File (SMF) ist ein zentraler Teil der Dokumentation in der pharmazeutischen Produktion, das Informationen über einen Produktionsstandort und dessen Anlagen enthält. Es wird vom Pharmahersteller erstellt, um die Einhaltung der Guten Herstellungspraxis (GMP) und behördlicher Standards zu sichern. Das SMF umfasst u.a. Standortdetails, Personalqualifikationen, Produktionsverfahren, Qualitätskontrollsysteme, Vertriebs- und Transportbedingungen sowie Zertifizierungen. Es dient als essentielles Dokument für Zulassungsbehörden, Kundenbewertungen und Inspektionen.
SOP – Standard Operation Procedure (Standard-Arbeitsanweisung)
Dokumentierte Arbeitsanweisung, die regelmäßig ausgeführte Prozesse und Aufgaben detailliert beschreibt. Diese Anweisungen dienen dazu, die Konsistenz und Qualität bei der Durchführung von Prozessen zu gewährleisten. SOPs sind entscheidend, um sicherzustellen, dass alle Mitarbeiter die erforderlichen Standards und Vorschriften einhalten und dadurch die Produktqualität, Sicherheit und regulatorische Konformität bewahrt wird.
Steam Quality (Dampfqualität)
Die Dampfqualität bei der Sterilisation bezeichnet die spezifischen Eigenschaften des Dampfes, die für eine erfolgreiche Sterilisation erforderlich sind. Es definiert das Massenverhältnis von Dampf zu Wasser in einem Dampf-Wasser-Gemisch.
Folgende Dampfeigenschaften sind für die Sterilisation von Bedeutung:
- Hoher Energiegehalt für optimale Wärmeübertragung und Erreichen der Sterilisationstemperaturen.
- Erhöhtes Durchdringungsvermögen, das eine vollständige Abdeckung der zu sterilisierenden Gegenstände gewährleistet.
- Fähigkeit zur Kondensation und Verdrängung von Luft aus der Kammer, was eine effektive Sterilisation ermöglicht.
Minderwertige Dampfqualität kann die Sterilisation beeinträchtigen, da sowohl die Wärmeübertragung als auch die Dampfdurchdringung behindert werden können. Daher ist die Überwachung und Kontrolle der Dampfqualität in der Sterilisation mit feuchter Hitze von entscheidender Bedeutung.
SVPs – Small Volume Parenterals
SVPs, oder "Small Volume Parenterals", beziehen sich auf sterile Injektionslösungen, die gewöhnlich in Mengen von <100 ml abgefüllt werden. Diese Lösungen werden in unterschiedlichen Behältern verpackt, abhängig von ihrem vorgesehenen Verwendungszweck. SVPs werden häufig zur Verabreichung von Medikamenten eingesetzt und müssen strengen Sterilisationsanforderungen entsprechen.
Temperature (Heat) Penetration Study (Temperatur-Penetrationsstudie)
Der Zweck einer Temperatur-Penetrationsstudie besteht darin, das Heiz- und Kühlverhalten einer Produkt-/Verpackungskombination in einem spezifischen Sterilisationsverfahren zu bestimmen, um sichere thermische Prozesse festzulegen und Prozessabweichungen zu bewerten. Die Studie muss so gestaltet sein, dass sie alle kritischen Faktoren, die die Heizraten beeinflussen und mit dem Produkt, der Verpackung und dem Prozess zusammenhängen, angemessen und genau untersucht. Oftmals werden Penetrationsstudien parallel zu Temperaturverteilungsstudien durchgeführt.
Temperature Mapping Study (Temperaturverteilungsstudie)
Temperaturverteilungsstudien finden in verschiedenen GMP-Bereichen sowie in unterschiedlichen Richtlinien und Normen Anwendung. Sie werden überall dort durchgeführt, wo die Temperatur entscheidend für die Prozessqualität und letztlich die Qualität des Endprodukts ist. Dazu gehören unter anderem Sterilisationsprozesse, Kühl- und Gefrieranlagen, Lagerräume, Stabilitätskammern sowie Transportbehälter im Bereich der Kühlkette. So wird beispielsweise in einem Dampfautoklaven der sogenannte Cold-Spot und Hot-Spot ermittelt, während das Temperatur-Mapping in einem Lagerraum unter anderem die spätere Platzierung fest installierter Überwachungssensoren unterstützt.
Traceability (Rückführbarkeit, Rückverfolgbarkeit)
Rückführbarkeit bedeutet generell, alle Schritte eines Produkts oder Prozesses einer Charge oder auch durchgeführter Maßnahmen von der Herkunft der Rohmaterialien bis zur endgültigen Verteilung des Fertigproduktes nachvollziehen zu können. Rückführbarkeit z.B. bei der Kalibrierung eines Temperatursensors bedeutet, dass die Kalibrierung und Justage auf ein nationales Temperaturnormal zurückgeführt werden kann.
URS – User-Requirements-Specifications (Benutzerspezifikationen)
Dies ist ein Dokument, das die Anforderungen und Erwartungen des Auftraggebers an ein neues Produkt oder System beschreibt. Es enthält, was das System leisten soll, und bildet zusammen mit den technischen und regulatorischen Anforderungen die Grundlage für das sogenannte Lastenheft. Das Lastenheft wird in der Regel vom Kunden oder Auftraggeber erstellt.
Dieses Dokument ist ein Baustein des V-Modells. Es fasst Anforderungen aus verschiedenen Quellen zusammen und unterstützt das Design, die Inbetriebnahme, die Qualifizierung sowie den Betrieb und die Wartung eines Systems. Kritische Qualitätsmerkmale und Prozessparameter sind wichtige Bestandteile, die zur Unterstützung des qualitätsrisikobasierten Prozesses identifiziert werden müssen. Die Spezifikationen können flexibel gestaltet werden, um die Anforderungen eines Mehrzweckbetriebs zu erfüllen, und sind dynamische Dokumente, die bei Änderungen aktualisiert werden.
USP® – United States Pharmacopeia (Arzneibuch USA)
Die United States Pharmacopeia (USP®) ist eine US-amerikanische Non-Profit-Organisation, die Standards für die Qualitätssicherung von Arzneimitteln und Nahrungsergänzungsmitteln entwickelt. Diese Standards sind wichtig für die Einhaltung von Vorschriften in der pharmazeutischen Industrie weltweit.
Validation (Validierung)
Validierung ist der dokumentierte Prozess zur Überprüfung, dass ein System, Prozess, Gerät oder Produkt die spezifischen Anforderungen und Qualitätsstandards zuverlässig erfüllt. Dieser Prozess stellt sicher, dass alle kritischen Parameter kontrolliert werden und konsistent reproduzierbare Ergebnisse erzielt werden, um sowohl die Sicherheit als auch die Wirksamkeit des Endprodukts zu gewährleisten. Somit ist die Validierung der dokumentierte Nachweis, dass alle Verfahren, Prozesse, Anlagen, Systeme, Materialien und Geräte mit den GxP-Vorgaben übereinstimmen und die erwarteten Ergebnisse liefern.

Validation Matrix (Validierungsmatrix)
Besonders bei komplexen Validierungen erleichtert die Validierungsmatrix den Überblick. Sie ist ein strukturiertes Dokument oder Tool, das die Beziehung zwischen verschiedenen Validierungsanforderungen und den zu validierenden Elementen, wie Systemen, Prozessen oder Geräten, darstellt. Diese Matrix hilft dabei sicherzustellen, dass alle Aspekte eines Projekts den regulatorischen und unternehmensinternen Standards entsprechen.
Typischerweise enthält eine Validierungsmatrix Informationen zu:
- Validierungsanforderungen: Spezifische Kriterien, die erfüllt werden müssen, basierend auf regulatorischen Richtlinien und Unternehmensstandards.
- Zu validierende Elemente: Auflistung der Systeme, Prozesse oder Komponenten, die validiert werden müssen.
- Tests und Prüfkriterien: Methoden und Verfahren, die angewendet werden, um die Erfüllung der Anforderungen zu bestätigen.
- Dokumentation: Referenzen zu den Dokumenten, die die Ergebnisse und die Nachweise der Validierung enthalten.
- Verantwortlichkeiten: Zuweisung von Aufgaben an spezifische Personen oder Teams für die Durchführung der Validierungsaktivitäten.
Validation Plan (Validierungsplan)
Die Validierungsplanung ist ein wesentlicher Prozess in der Qualitätssicherung, bei dem überprüft wird, ob ein System oder Produkt den festgelegten Anforderungen entspricht. Um dies zu erreichen, werden spezifische Tests und Prüfungen geplant und dokumentiert. Neben der Planung ist häufig auch eine Risikoanalyse erforderlich, um mögliche Schwachstellen zu identifizieren und gezielte Validierungsmaßnahmen zur Risikominderung zu entwickeln.
In vielen Branchen, insbesondere in der Pharma-, Medizin- und Lebensmittelindustrie, treibt die Einhaltung regulatorischer Anforderungen die Validierungsplanung. Es ist entscheidend, dass alle Tests und Prüfungen diesen Vorschriften entsprechen, um Compliance zu gewährleisten. Eine gründliche Dokumentation des Validierungsprozesses ist unerlässlich, um die Einhaltung der Qualitätsanforderungen nachzuweisen, insbesondere bei Audits oder Inspektionen.
Validation Protocol (Validierungsprotokoll)
Ein Validierungsprotokoll ist eine detaillierte, schriftliche Zusammenfassung, die spezifische Tests und Verfahren zur Überprüfung beschreibt, ob ein Produkt die Anforderungen für seinen vorgesehenen Zweck erfüllt. Es enthält Anweisungen zur Durchführung der Validierung, Akzeptanzkriterien und die notwendige Dokumentation der Ergebnisse. Das Protokoll ist fokussierter als der Validierungsplan und konzentriert sich auf die praktische Umsetzung der Validierung für bestimmte Prozesse oder Produkte.
Validation Report (Validierungsbericht)
Nachdem die Validierungsaktivitäten abgeschlossen sind, dokumentiert der Validierungsbericht die Ergebnisse und bewertet, ob die Validierungsanforderungen erfüllt wurden. Der Bericht beinhaltet die Analyse und Interpretation der Daten, zeigt auf, ob die Akzeptanzkriterien erreicht wurden, und gibt Empfehlungen für zukünftige Maßnahmen.
VMP – Validation Master Plan
Der Validierungs-Masterplan (VMP) ist ein strategisches Dokument, das alle Validierungsaktivitäten einer Produktionsstätte leitet, um die konsistente Produktqualität und die Einhaltung von Qualitätsstandards sicherzustellen. Als wesentliches Element des Qualitätsengagements eines Unternehmens stellt der VMP sicher, dass Produkte und Prozesse systematisch validiert werden. Er unterstützt die Konsistenz und Kontrolle in der Produktion, minimiert Risiken und gewährleistet die regulatorische Compliance durch detaillierte Dokumentation und Anpassung an aktuelle Vorschriften. Zudem optimiert der VMP die Nutzung von Ressourcen und gibt klare Anleitungen zur Fehlerminimierung. Er muss dabei die geltenden regulatorischen Anforderungen, wie die der FDA und der EU, einhalten. Generell legt der VMP die Validierungsstrategie eines Unternehmens fest und definiert sowie dokumentiert die Absichten, Verantwortungsbereiche, Vorgehensweisen und Schlüsselaspekte eines Validierungsprogramms.
WFI – Water-for-Injection (Wasser für Injektionszwecke)
Wasser für Injektionszwecke (WFI) ist eine hochreine Form von Wasser, die speziell zur Herstellung von Arzneimitteln für die parenterale Verabreichung verwendet wird. Es dient als Trägermaterial zum Auflösen oder Verdünnen von Substanzen oder Zubereitungen, die in den Körper injiziert werden. WFI muss strenge Qualitätsanforderungen erfüllen, um sicherzustellen, dass es frei von Verunreinigungen und mikrobiologischen Kontaminationen ist. In der pharmazeutischen Industrie ist es ein wesentlicher Bestandteil zur Sicherstellung der Sicherheit und Wirksamkeit von injizierbaren Präparaten.
WHO – World Health Organization (Weltgesundheitsorganisation)
Die Weltgesundheitsorganisation (WHO), 1948 gegründet und mit Hauptsitz in Genf, fungiert als Koordinationsbehörde für internationale öffentliche Gesundheitsangelegenheiten. Ihr Ziel ist es, weltweit eine bessere und gesündere Zukunft für alle Menschen zu schaffen.
WIP – Washing-in-Place (Reinigung vor Ort)
WIP (Washing In Place) bezeichnet die teilautomatisierte Reinigung und Desinfektion von z.B Abfüllmaschinen durch das Einpumpen geeigneter Reinigungs- und Desinfektionsmittel in das System. Im Unterschied zu CIP (Cleaning-In-Place) erfordert WIP jedoch zusätzlichen manuellen Reinigungsaufwand.
Z-Value (Z-Wert)
Der Z-Wert ist ein mikrobiologischer Parameter, der das Abtötungsverhalten von Mikroorganismen bei der Sterilisation charakterisiert. Er beschreibt die notwendige Temperaturerhöhung, um den D-Wert um den Faktor zehn zu senken, was eine log-Zyklus-Reduktion der Mikroorganismen bewirkt. Der Z-Wert zeigt die Empfindlichkeit der Mikroben auf Temperaturänderungen und ist zentral, um eine effektive Mikroorganismenreduktion bei minimaler Produkteinwirkung zu gewährleisten.
ZLG – Zentralstelle der Länder für Gesundheitsschutz bei Arzneimitteln und Medizinprodukten
Die Zentralstelle der Länder für Gesundheitsschutz bei Arzneimitteln und Medizinprodukten (ZLG) ist eine deutsche Institution, die die Zusammenarbeit zwischen den Bundesländern in Fragen des Gesundheitsschutzes koordiniert. Sie spielt eine wichtige Rolle bei der Harmonisierung und Überwachung von Standards für Arzneimittel und Medizinprodukte in Deutschland. Die ZLG unterstützt die Länderbehörden bei der Überwachung, Zulassung und Zertifizierung von Produkten und Prozessen im medizinischen Bereich, um die Sicherheit und Wirksamkeit zu gewährleisten. Durch diese koordinierte Herangehensweise trägt die ZLG dazu bei, hohe Qualitätsstandards im Gesundheitsschutz sicherzustellen.