GxP Dictionary
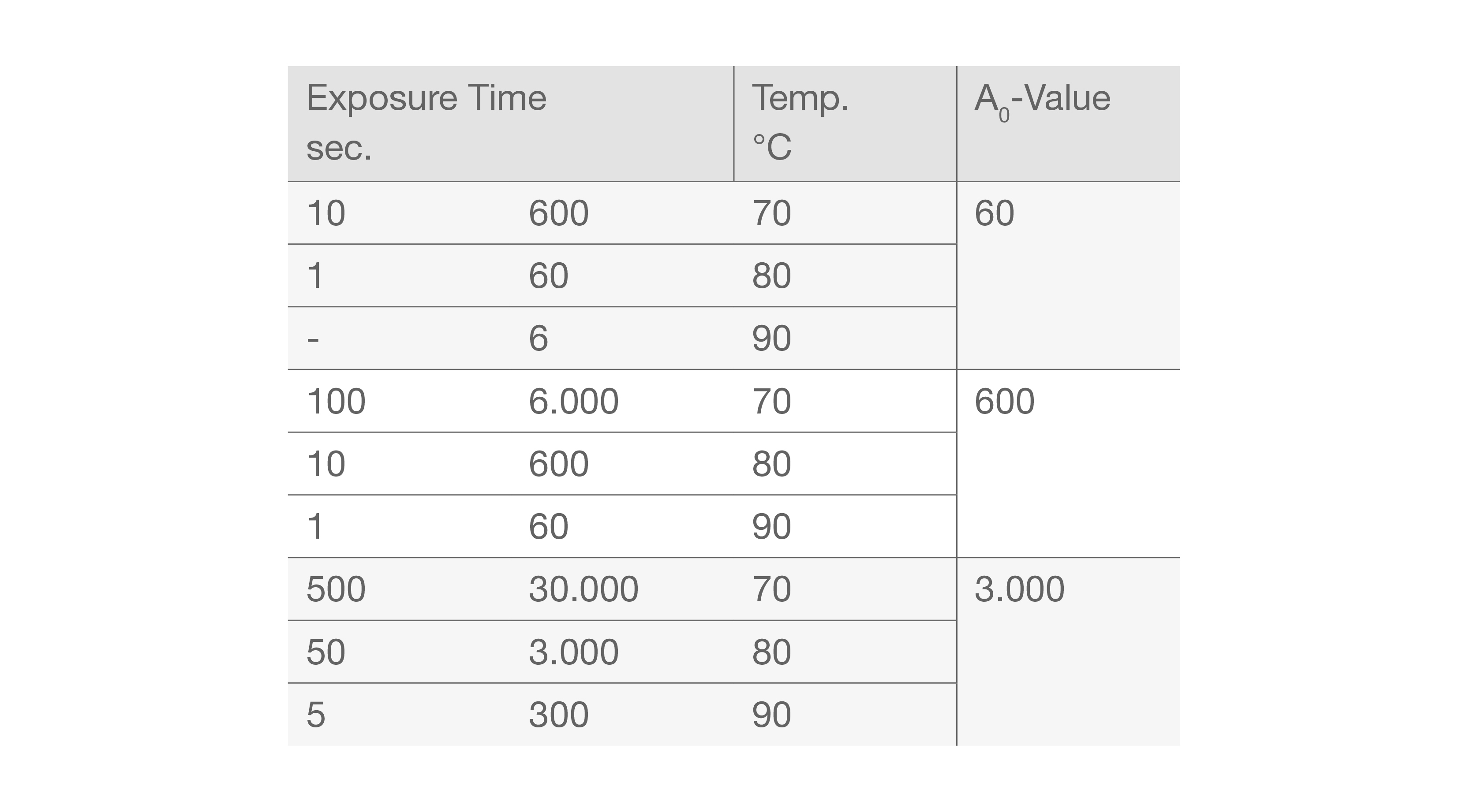
A0-Value
La norme ENDIN ISO 15883-1 définit la valeur A0 comme une mesure de l'inactivation microbienne lors de la désinfection par la chaleur humide. Cette valeur sert à déterminer la quantité de chaleur humide requise dans les lave-désinfecteurs automatisés. La valeur A0 représente le temps nécessaire pour obtenir une réduction spécifique de logarthmitrophes des micro-organismes basée sur la température du cycle de désinfection.
Principe ALCOA (++)
Le principe ALCOA vise à garantir l'intégrité et la qualité des données dans les enregistrements. ALCOA signifie :
- Attribuable - Les données doivent être traçables à une source ou une personne spécifique.
- Lisible - Les données doivent être lisibles et permanentes.
- Contemporain - Les données doivent être enregistrées au moment de l'activité ou de l'observation.
- Original - Les données doivent être dans leur forme originale, non modifiée.
- Précis - Les données doivent être exactes et sans erreur.
Le principe ALCOA++ étend le concept ALCOA original pour considérer des attributs supplémentaires de l'intégrité des données dans des environnements réglementés tels que l'environnement GxP. Cette extension inclut d'autres critères :
- Complet - Toutes les données doivent être complètes et exhaustives.
- Cohérent - Les données doivent être dans un format fiable et logique.
- Durable - Les données doivent être préservées au fil du temps.
- Disponible - Les données doivent être accessibles dès que nécessaire.
Annex 1
Depuis août 2023, la nouvelle Annexe 1 est entrée en vigueur dans une version révisée, assortie d'une période de transition s'étendant jusqu'en 2024. Si la structure fondamentale demeure inchangée, l'ajout de sous-chapitres supplémentaires a rendu cette Annexe plus exhaustive et détaillée. L'accent est mis tout particulièrement sur la production de produits stériles, afin de minimiser les risques de contamination microbiologique, particulaire et pyrogène. Par ailleurs, la nouvelle Annexe 1 fait office de guide pour assurer la meilleure protection possible des produits stériles.
API - Ingrédient Pharmaceutique Actif
Un ingrédient pharmaceutique actif (API) est la principale substance active pharmacologique d'un médicament qui atteint l'effet thérapeutique escompté. Les API sont des composés chimiques hautement purs incorporés dans des processus de fabrication stricts afin d'en garantir la qualité, la sécurité et l'efficacité. Ils sont souvent combinés avec des excipients dans les médicaments pour améliorer la forme posologique et la stabilité du produit final.
APR – Annual Product Review
L'examen annuel des produits (APR) exigé par la FDA est un processus obligatoire pour garantir la qualité des produits pharmaceutiques fabriqués ou importés aux États-Unis. L'APR implique une revue rétrospective et systématique de tous les lots, procédés de production, écarts, plaintes, ainsi que de toute modification des procédures de fabrication et des méthodes de contrôle dans l'année menée.
Audit
Dans un environnement GxP, un audit désigne un examen systématique et indépendant sur site réalisé afin de s'assurer que tous les processus, systèmes et procédures respectent les réglementations et normes de qualité applicables. Les audits visent à garantir l'intégrité et la qualité des produits en identifiant les faiblesses et les risques et en évaluant l'efficacité des mesures de contrôle qualité. Les audits peuvent être réalisés par diverses entités, tant en interne qu'en externe, comme par les clients. Distincte dans leur terminologie, mais ayant le même objectif sous-jacent, est une inspection, qui ne peut être menée que par les autorités de régulation compétentes.
Audit Trail
Le terme « trace d'audit » désigne un enregistrement organisé de tous les changements et actions au sein d'un système ou d'un document. Cette journalisation garantit l'intégrité et la traçabilité des données, qui sont essentielles pour respecter les normes réglementaires. La trace d'audit permet de clarifier qui a apporté des modifications, quand et pourquoi, en faisant un outil essentiel pour l'assurance qualité. Avec l'essor de la numérisation, ce concept a pris de l'importance en matière de sécurité et d'intégrité des données, et il est explicitement mentionné dans des réglementations telles que 21 CFR Partie 11, ALCOA++ ou l'Annexe GMP 11 de l'UE.
Bioburden
Décrivez le nombre total de micro-organismes viables présents sur une surface non stérilisée ou dans le produit.
BP – British Pharmacopoeia
La Pharmacopée britannique (BP) est le recueil officiel des normes médicales au Royaume-Uni, fournissant des normes complètes pour garantir la qualité et la sécurité des médicaments. Il sert de référence important pour les fabricants, les laboratoires d'essais et les autorités de réglementation.
21 CFR - Code des règlements fédéraux
Le Code des règlements fédéraux (CFR) est une collection systématique des règles générales et permanentes publiées dans le Federal Register par les départements et agences du gouvernement fédéral des États-Unis. Le Titre 21 du CFR est spécifiquement dédié aux réglementations de la Food and Drug Administration (FDA) et traite des aspects liés à la régulation des aliments et médicaments.
21 CFR partie 11
21 CFR Partie 11 est un règlement de la Food and Drug Administration (FDA) des États-Unis qui définit les critères d'acceptation des dossiers électroniques et des signatures électroniques. Elle précise les exigences concernant l'intégrité, la sécurité et la disponibilité de ces documents afin de garantir qu'ils soient aussi fiables et fiables que les documents traditionnels sur papier.
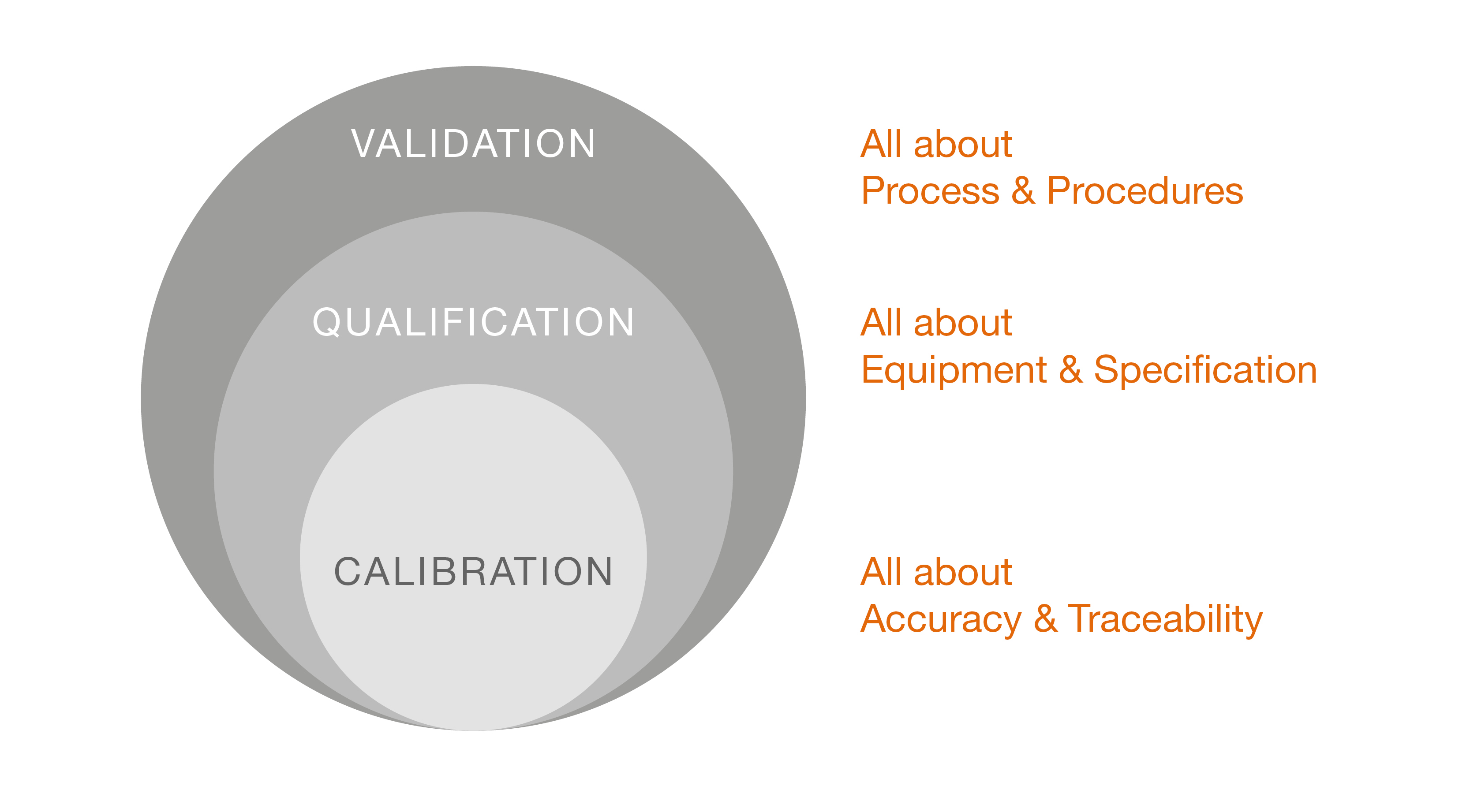
Calibration
L'étalonnage est la comparaison documentée d'un dispositif de mesure à calibrer avec une référence traçable. Tous les capteurs et dispositifs de mesure doivent être calibrés avant d'être utilisés dans une étude de validation. Les résultats d'étalonnage doivent être documentés. Les incertitudes de mesure réalisables doivent être alignées sur les exigences spécifiques à chaque application.
- Selon le guide ISO/IEC 99:2007, l'étalonnage est un processus qui établit une relation entre les grandeurs de mesure et les indications correspondantes.
- Étalonnage = différence déterminée entre la valeur mesurée et la vraie valeur (référence/étalon traçable).
- Ajustement = correction de la valeur mesurée vers la valeur réelle afin de minimiser l'erreur de mesure et d'augmenter la précision.
- L'étalonnage et l'ajustement ont lieu tout au long de la durée de vie d'un système, d'un instrument, d'un capteur, et doivent être effectués.
- Cela fait partie de la qualification et de la validation mais ne les remplace pas.
- Il comprend tous les capteurs et grandeurs de mesure, instruments ou un système, et définit la précision globale de l'ensemble de la chaîne de mesure.
- Il fait référence aux performances et caractéristiques des capteurs, des systèmes de mesure et des logiciels.
- Elle comprend des méthodes générales de métrologie :
- Définition de la méthode de mesure (conditions environnementales ; méthode des normes requises)
- Création d'un modèle mathématique pour l'évaluation de l'étalonnage, incluant son incertitude de mesure
- Performance de l'étalonnage
- Création d'un certificat d'étalonnage avec des détails (déviation déterminée, ajustement et incertitude de mesure)
- Selon le procédé/capteur/quantité de mesure, une post-étalonnage sans ajustement est nécessaire pour la vérification et la documentation après utilisation.
Calibration Protocol
Un protocole d'étalonnage est un document technique qui décrit un plan complet ou une procédure opérationnelle standard pour la réalisation de l'étalonnage d'un dispositif, d'un instrument ou d'un système. Il comprend toutes les étapes et conditions nécessaires pour garantir que l'appareil fournit des mesures précises et fonctionne dans les tolérances spécifiées. Un protocole d'étalonnage établit des exigences spécifiques d'étalonnage, incluant les méthodes à utiliser, les intervalles d'étalonnage, les limites acceptables et la documentation nécessaire des résultats.
CAPA – Corrective and Preventive Action
C'est une approche systématique permettant d'identifier et de traiter les causes profondes des écarts et de mettre en place des mesures préventives pour prévenir leur récidive. Un système robuste de CAPA (Action corrective et préventive) comprend l'analyse des causes profondes, la mise en œuvre d'actions correctives, l'évaluation de l'efficacité de ces actions et, bien sûr, une documentation complète accompagnée.
CC – Change Control
Le contrôle des changements est un processus systématique et formel qui garantit que toutes les modifications apportées aux produits, procédés ou systèmes sont soigneusement planifiées, évaluées et documentées. L'objectif principal du contrôle des changements dans l'environnement des BPF (bonnes pratiques de fabrication) est de préserver l'intégrité et la qualité des produits malgré les changements, et de veiller à ce que ces modifications respectent les exigences réglementaires.
CFR – Code Federal Regulations
Collecte des règles générales et permanentes émises par les agences fédérales des États-Unis. Dans les industries pharmaceutique et biotechnologique, le 21 CFR joue un rôle particulièrement central, car il contient les réglementations de la Food and Drug Administration (FDA) des États-Unis afin d'assurer la sécurité, l'efficacité et la qualité des produits pharmaceutiques et des dispositifs médicaux.
CFU – Colony Forming Unit
L'unité de formation de colonies (CFU) est une unité de mesure utilisée en microbiologie pour estimer le nombre de micro-organismes viables dans un échantillon. Les UFC sont essentielles pour évaluer la contamination microbiologique des produits et des matériaux. Une unité formatrice de colonies est définie comme un micro-organisme viable, tel que des bactéries ou des champignons, qui peut se multiplier et former plusieurs colonies pendant la culture.
ChP – Chinese Pharmacopoeia
La Pharmacopée chinoise (ChP), également connue sous le nom de PPRC, est le compendium officiel de la République populaire de Chine et comprend des normes complètes concernant la pureté, la description, les tests, le dosage, les mesures de sécurité, le stockage et la puissance des médicaments. Elle joue un rôle crucial dans la garantie de la qualité des médicaments en Chine, à l'instar de la Pharmacopée européenne en Europe et de la USP aux États-Unis.
CIP – Clean in Place
Il s'agit d'une procédure permettant de nettoyer les équipements et systèmes de production sans démontage. Le CIP (Cleaning in Place) est utilisé dans l'industrie pharmaceutique pour nettoyer des tuyaux, des contenants, des systèmes de remplissage ou des réservoirs, par exemple. La méthode consiste en une combinaison de rinçage, de nettoyage et de désinfection avec divers agents nettoyants. Souvent, la stérilisation est ensuite réalisée à l'aide de ce qu'on appelle SIP (Steam in Place).
Cold Chain
Une chaîne du froid est un processus d'approvisionnement à température contrôlée dans lequel des produits sensibles à la température tels que les aliments, les produits pharmaceutiques, les substances chimiques et les matériaux biologiques sont transportés et stockés dans une plage de température spécifiée tout au long de leur chaîne d'approvisionnement. L'objectif est d'assurer la qualité, la sécurité et l'efficacité de ces produits en respectant strictement les exigences de température spécifiées. La chaîne du froid englobe l'ensemble du processus, de la production au stockage, au transport et à la distribution jusqu'au consommateur final.
Dans le domaine pharmaceutique, cela inclut les médicaments, les ingrédients actifs pharmaceutiques (API), les vaccins et les substances biologiques. Ces produits sont très sensibles aux fluctuations de température, et toute déviation de la plage recommandée peut les rendre inefficaces voire dangereux.
Cold Chain Management
La gestion de la chaîne du froid désigne le contrôle et la surveillance complets de toutes les phases d'une chaîne d'approvisionnement à température contrôlée afin d'assurer l'intégrité des produits sensibles à la température tels que les aliments, les produits pharmaceutiques et les matériaux biologiques. Cela englobe l'ensemble du processus, de la production au stockage et au transport, jusqu'à la livraison finale au consommateur final. Une gestion efficace de la chaîne du froid garantit que ces produits restent efficaces et sûrs et que toutes les exigences réglementaires pertinentes sont respectées.
Compliance
Dans les environnements réglementés, la conformité fait référence au respect de toutes les exigences et normes légales, éthiques et réglementaires pertinentes. Dans des secteurs tels que la pharmacie, l'alimentation et la technologie médicale, cela signifie s'assurer que tous les procédés, systèmes et produits respectent les réglementations établies et les meilleures pratiques afin de garantir la sécurité, l'efficacité et la qualité. En fin de compte, il s'agit d'assurer la sécurité des patients.
Concurrent Validation
La validation qui l'accompagne, également appelée validation concurrente, a lieu parallèlement à la production régulière en cours. Son objectif est d'évaluer la performance et la cohérence du processus de fabrication et de s'assurer que les exigences de qualité et réglementaires sont toujours respectées.
CPV – Continued Process Validation
La validation continue des procédés (CPV) garantit que les procédés et composants de production restent systématiquement dans les limites de qualité établies et est considérée comme la troisième phase de validation des procédés. Le CPV vise à maintenir les processus sous contrôle continu et à maintenir les normes de qualité grâce à un suivi régulier. Un CPV efficace identifie les incohérences des processus et permet des actions correctives rapides.
CQA – Critical Quality Attributes
Les Attributs Critiques de Qualité (CQA) sont des propriétés ou caractéristiques physiques, chimiques, biologiques ou microbiologiques spécifiques d'un produit qui doivent respecter des limites, plages ou distributions spécifiées afin de garantir que la qualité souhaitée soit atteinte. Ces attributs sont essentiels à la fabrication et à l'assurance d'un niveau constant de qualité produit.
CSV – Computer-System/ -Software-Validation
La description fait référence à la validation des systèmes informatiques (CSV), un processus critique visant à garantir qu'un système ou un logiciel informatique fonctionne comme prévu et sans erreur, en particulier dans les environnements soumis aux directives actuelles de bonnes pratiques de fabrication (cGMP). Le processus CSV comprend des exigences telles que l'analyse des risques, la planification, les tests et la validation, la documentation et le suivi continu. Une validation adéquate des systèmes informatisés est essentielle pour maintenir l'assurance qualité et la conformité.
D-Value
La valeur D, ou temps de réduction décimal, est le temps en minutes nécessaire pour réduire la population microbienne de 90 %, correspondant à une réduction d'un log. Lors de la stérilisation à la vapeur, la valeur D mesure l'efficacité de tuée du procédé. Il est crucial pour le développement et la validation des procédures de stérilisation, car il garantit une réduction de la charge microbienne sans compromettre l'intégrité du produit.
Data Integrity
L'intégrité des données fait référence à la complétude, à la justesse, à la cohérence et à la précision des données tout au long de leur cycle de vie. Elle est essentielle pour la fiabilité des données utilisées dans des industries réglementées comme la pharmacie et la technologie médicale. L'intégrité des données garantit que les données sont fiables et traçables, ce qui est essentiel pour la conformité réglementaire ainsi que pour la sécurité et l'efficacité des produits. C'est un élément clé du système d'assurance qualité et il est imposé par de nombreuses directives et normes comme fondement de la conformité à l'assurance qualité.
Design Specifications
Les spécifications de conception font référence à des documents techniques détaillés qui décrivent les exigences et les caractéristiques d'un système ou d'un produit. Ils définissent comment un système ou un composant doit fonctionner pour répondre à certaines normes et exigences réglementaires. Ces spécifications servent de base au développement et à la validation des systèmes, garantissant que tous les aspects pertinents tels que la fonctionnalité, la sécurité et la conformité sont pris en compte.
Deviation
Dans l'environnement technique pharmaceutique, le terme « déviation » désigne un écart par rapport aux normes, procédures ou spécifications établies au sein d'un procédé réglementé, tel que la fabrication, les tests ou le contrôle qualité des produits pharmaceutiques. Les écarts peuvent être attribués à des erreurs de procédé, des erreurs humaines ou des événements imprévus et doivent être documentés, enquêtés et corrigés de manière approfondie et méticuleuse afin d'assurer la sécurité et l'efficacité des produits.
Deviation Management
La gestion des écarts désigne le processus structuré permettant d'identifier, documenter, enquêter et corriger les écarts survenus lors de la fabrication, des tests ou du contrôle qualité des produits pharmaceutiques. Ce processus est crucial pour garantir l'intégrité et la qualité des produits ainsi que la conformité aux exigences réglementaires.
La gestion des déviations comprend les étapes suivantes :
- Détection : Identification d'une déviation par rapport à la procédure ou à la spécification standard.
- Documentation : Document écrit de la déviation, y compris ses détails et ses impacts potentiels.
- Investigation : Analyse des causes de la déviation afin d'en déterminer l'origine et les raisons.
- Actions correctives : mise en œuvre des actions visant à traiter les impacts immédiats de la déviation.
- Mesures préventives : Introduction de mesures pour prévenir la réapparition de la déviation.
- Revue et clôture : Évaluation de l'efficacité des actions entreprises et fermeture formelle de la déviation.
Une gestion efficace des écarts est essentielle pour maintenir la conformité réglementaire et améliorer continuellement la qualité des produits.
DMS – Document Management System
Un système de gestion documentaire (SGD) est une solution logicielle qui organise, stocke et gère les documents électroniques au sein d'une entreprise. Il permet aux utilisateurs de créer, modifier, stocker et récupérer efficacement des documents tout en surveillant simultanément les droits d'accès et en modifiant les journaux. Dans une grande organisation, le DMS aide à optimiser le flux d'informations, à répondre aux exigences de conformité et à renforcer la collaboration entre différents domaines métier. Il peut gérer à la fois des données structurées, telles que des rapports et des politiques, et du contenu non structuré, comme les emails et le multimédia, sur une plateforme centrale et accessible.
DQ – Design Qualification
La qualification de conception (DQ) est la vérification documentée que les documents de conception créés respectent les exigences utilisateur spécifiées dans la spécification des exigences. Il confirme également que tous les documents de conception nécessaires à la mise en œuvre ont été préparés, en tenant compte des exigences GMP (lois, directives et état de l'art).
EMA – European Medicines Agency
L'EMA, ou Agence européenne des médicaments, est une agence de l'Union européenne responsable de l'évaluation scientifique, de la supervision et du suivi de la sécurité des médicaments dans l'UE. Elle joue un rôle central dans l'approbation de nouveaux médicaments afin de garantir qu'ils respectent des normes élevées de qualité, de sécurité et d'efficacité. En évaluant et en surveillant les médicaments au sein de l'Union européenne (UE) et de l'Espace économique européen (EEE), l'EMA protège et promeut la santé des personnes et des animaux.
EP – or Ph.Eur European Pharmacopeia
La Pharmacopée européenne (Ph. Eur.) est la principale source de normes officielles de qualité pour les médicaments et leurs ingrédients en Europe. La Direction européenne pour la qualité des médicaments et des soins de santé (EDQM), une direction du Conseil de l'Europe, assure un soutien scientifique et administratif pour celle-ci. L'organe directeur de la Pharmacopée européenne est la Commission de la Pharmacopée européenne, qui élabore et fixe les normes.
EU-GMP – Annex
Dans le cadre des lignes directrices de l'UE sur les BPF (Bonnes pratiques de fabrication), les annexes fournissent des documents complémentaires qui proposent des réglementations et des lignes directrices spécifiques pour des domaines particuliers des BPF. Alors que la partie principale des directives GMP couvre les exigences générales relatives aux procédés de production et au contrôle qualité dans la fabrication pharmaceutique, les annexes abordent des sujets particuliers ou des formes spécialisées de fabrication.
Chaque annexe se concentre sur un sujet spécifique différent, comme la fabrication de produits stériles, les procédures pour certaines applications technologiques ou l'examen d'aspects spécifiques de la qualité. Ces annexes aident les fabricants à mieux comprendre et à répondre aux défis et exigences uniques dans divers domaines. Actuellement, des annexes sont disponibles de l'annexe 1 à l'annexe 20.
EU-GMP Annex 11 – Computerized Systems
L'annexe a été révisée pour répondre à l'utilisation accrue et à la complexité croissante des systèmes informatisés employés dans les opérations réglementées par les BPF. Ces systèmes comprennent des composants logiciels et matériels travaillant ensemble pour remplir des fonctions spécifiques. Ils doivent être validés et l'infrastructure informatique qualifiée. Lorsque les activités manuelles sont remplacées par des systèmes informatisés, la qualité du produit, le contrôle des procédés et l'assurance qualité ne doivent pas être compromis, et le risque global du procédé ne doit pas augmenter.
EU-GMP Annex 15 – Qualification and Validation
Ce document fournit des indications sur l'interprétation des principes BPF pour les produits médicinaux humains et vétérinaires conformément aux directives 2003/94/CE et 91/412/CEE. Il décrit la qualification et la validation des installations, équipements, utilités et procédés dans la fabrication de médicaments, et il peut être appliqué en option aux substances actives sans imposer d'exigences supplémentaires. Les fabricants doivent contrôler les aspects critiques tout au long du cycle de vie du produit et du procédé grâce à la qualification et à la validation. Les changements affectant la qualité du produit doivent être documentés, et leur impact sur le statut de validation doit être évalué. Les systèmes informatisés doivent être validés conformément à l'Annexe 11, en tenant compte des directives ICH Q8, Q9, Q10 et Q11.
EU-GMP Annex 20 – Quality Risk Management (QRM)
L'annexe 20 des lignes directrices GMP de l'UE, qui s'alignent sur la ligne directrice ICH Q9, fournit des orientations pour la gestion systématique des risques de qualité (QRM). Elle aide à garantir la conformité aux exigences GMP et autres normes de qualité en fournissant des principes et des méthodes pour la gestion formelle des risques de qualité.
EU-GMP Guidelines
La Commission européenne a fixé des exigences en matière d'assurance qualité dans les processus et environnements de production selon les principes de bonnes pratiques de fabrication (BPF) pour les produits médicinaux destinés à l'usage humain afin d'assurer la vérification des procédés. Des directives détaillées pour l'interprétation de ces principes BPF se trouvent dans le Guide des BPF de l'UE :
- Guide des GMP de l'UE Partie I : Guide des bonnes pratiques de fabrication.
- Guide GMP de l'UE Partie II : Exigences de base pour les substances actives utilisées comme matières de départ.
- Guide GMP de l'UE Partie III : Documents relatifs aux GMP.
- Guide GMP de l'UE Partie IV : exigences GMP pour les médicaments de thérapie avancée.
F0-Value
La valeur F0 mesure l'efficacité de la stérilisation à la vapeur, définie comme le temps en minutes nécessaire à 121,1°C pour détruire tous les micro-organismes, en tenant compte d'une valeur Z spécifiée. Ce concept est essentiel pour garantir que les processus de stérilisation soient rigoureux et cohérents, en maintenant la sécurité et l'efficacité des produits, en particulier dans des secteurs tels que la pharmacie et la technologie médicale.
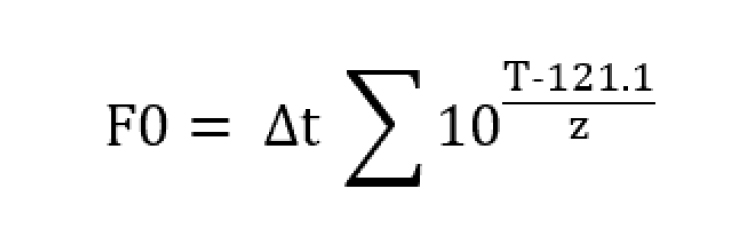
FAT – Factory-Acceptance-Test
Le test d'acceptation en usine (FAT) est un processus d'inspection et d'approbation d'un produit effectué dans l'usine du fabricant avant sa livraison au client. L'objectif du FAT est de vérifier la fonctionnalité et la qualité de l'équipement dans des conditions contrôlées afin de s'assurer qu'il répond aux exigences spécifiées. Après la livraison au client, un test d'acceptation sur site (SAT) est généralement réalisé, qui teste le produit à l'emplacement final du client afin de vérifier l'installation correcte et la préparation opérationnelle. Ces tests sont des étapes cruciales dans le processus de gestion de la qualité afin de garantir que le produit répond aux exigences et fonctionne de manière fiable.
FDA – 482
La FDA peut inspecter un établissement pour diverses raisons, telles que des enquêtes de routine, des enquêtes ou en réponse à des problèmes signalés. À leur arrivée sur le site, l'enquêteur présentera ses accréditations et le formulaire d'inspection (formulaire FDA 482).
https://www.fda.gov/industry/fda-basics-industry/what-should-i-expect-during-inspection
FDA – 483
Le formulaire FDA 483 est utilisé pour documenter les lacunes observées lors d'une inspection FDA. L'enquêteur le présente lors de la réunion de clôture de l'inspection. Après émission, le formulaire est également envoyé au bureau de district responsable, chargé d'évaluer l'inspection. Selon l'importance des lacunes notées, une lettre d'avertissement peut suivre comme prochaine mesure réglementaire. Les documents FDA 483 délivrés sont également rendus publics sur le site web de la FDA.
FDA – Food and Drug Administration
La FDA (Food and Drug Administration) est une agence du Département américain de la Santé et des Services sociaux responsable de la réglementation et de la supervision des aliments, des médicaments pharmaceutiques, des dispositifs médicaux, des vaccins, des produits biologiques, des cosmétiques et des produits du tabac. Cela garantit que ces produits sont sûrs, efficaces et correctement étiquetés. De plus, la FDA est responsable de l'approbation et du contrôle de ces produits, ainsi que des marchandises importées relevant de sa juridiction.
FDA – Warning Letter
Une lettre d'avertissement de la FDA est une notification formelle de la Food and Drug Administration (FDA) des États-Unis envoyée aux entreprises ayant enfreint les exigences légales ou réglementaires. La lettre met en lumière des infractions spécifiques identifiées lors d'une inspection ou d'un examen et exige que l'entreprise règle rapidement les problèmes. Recevoir une telle lettre est grave, car le non-respect du délai prévu pour corriger les lacunes pourrait entraîner le refus d'une approbation ou un arrêt complet de l'importation. Les lettres d'avertissement sont accessibles publiquement sur le site web de la FDA.
https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/compliance-actions-and-activities/warning-letters
FDA Guidance for Industry – Process Validation
Cette directive résume les principes et approches recommandés par la FDA pour la validation des procédés dans la fabrication de médicaments et de produits biologiques pour l'homme et les animaux. Il intègre les meilleures pratiques que les fabricants peuvent utiliser pour valider leurs processus de production et les aligne sur le concept du cycle de vie du produit ainsi que sur les directives existantes de la FDA et de l'ICH.
Fh-Value
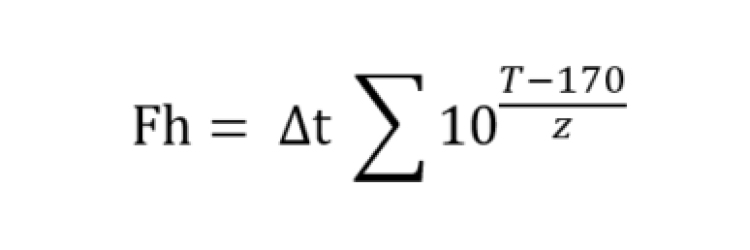
La valeur Fh, souvent appelée facteur de pénétration de chaleur dans le contexte de la stérilisation à la chaleur sèche, est un paramètre important pour évaluer l'efficacité et l'efficacité du procédé de stérilisation. Contrairement à la stérilisation à la chaleur humide, qui utilise généralement de la vapeur à 121,1°C, la stérilisation à la chaleur sèche utilise des températures plus élevées. La valeur Fh quantifie le temps et la température nécessaires pour atteindre un niveau spécifique de réduction microbienne dans ces conditions.
FMEA – Failure Mode and Effects Analysis
La FMEA, ou analyse des modes de défaillance et des effets (Failure Modes and Effects Analysis), est une méthode systématique permettant d'identifier et d'évaluer les défaillances potentielles d'un produit ou d'un procédé ainsi que leurs effets avant qu'elles ne surviennent. Il aide à identifier les risques pour mettre en œuvre des mesures proactives d'atténuation des risques. L'analyse évalue la gravité des défaillances potentielles, la probabilité de leur survenue et la détectabilité de ces défaillances. Cette méthodologie est largement utilisée en gestion de la qualité, notamment dans les domaines qui doivent répondre à des exigences élevées de sécurité et de fiabilité.
FMECA – Failure Mode, Effects, and Criticality Analysis
L'approche méthodique dont vous faites référence est FMECA, qui signifie Analyse du mode de défaillance, des effets et de la criticité. Ce processus s'appuie sur la FMEA en identifiant non seulement les modes de défaillance potentiels et leurs effets, mais aussi en évaluant la critique de ces modes de défaillance. L'analyse de la criticité aide à prioriser les risques en fonction de leur gravité, de leur occurrence et de leur détectabilité, permettant aux équipes de mettre en place des stratégies de mitigation des risques plus ciblées et efficaces.
FTA – Fault Tree Analysis
est une technique méthodologique permettant d'identifier et d'analyser les différents facteurs pouvant conduire à un événement de défaillance spécifique dans un système.
GAMP® – Good Automated Manufacturing Practice
Le guide GAMP®, publié pour la première fois en 1994 par le Pharmaceutical Industry Computer Systems Validation Forum (aujourd'hui le GAMP® Forum) en collaboration avec l'ISPE®, est devenu une norme pour la validation des systèmes informatisés dans l'industrie pharmaceutique. Bien qu'il ne soit pas juridiquement contraignant et que des approches alternatives de validation soient possibles, il constitue une référence importante pour les fabricants et les fournisseurs.
GAMP® 5V-Model
GAMP® 5 est une ligne directrice pour la validation des systèmes automatisés dans l'industrie pharmaceutique. Le V-Model représente le processus de développement et de validation en forme de V, chaque phase de développement correspondant à gauche à une phase de test spécifique à droite. Ce modèle permet de garantir que les systèmes sont rigoureusement testés et que les normes réglementaires sont respectées. En suivant une approche séquentielle, il garantit que toutes les étapes de documentation et de validation sont réalisées dans un ordre logique. Les responsabilités et responsabilités entre fournisseurs et clients peuvent également être définies au sein du V-Modèle. Ce faisant, elle capture non seulement les aspects techniques mais aussi organisationnels, ce qui facilite la collaboration entre les parties impliquées dans un projet.

GCP – Good Clinical Practice
Le GCP, ou Bonne Pratique Clinique, englobe des lignes directrices reconnues internationalement qui établissent des normes éthiques et scientifiques pour la planification et la conduite des essais cliniques. Ces directives garantissent que les droits, la sécurité et le bien-être des participants à l'essai sont protégés et que les données collectées durant les essais sont crédibles et exactes.
GDP – Good Distribution Practice
Comprend des mesures et des directives importantes pour la bonne distribution des produits pharmaceutiques. Ces pratiques sont conçues pour maintenir la qualité et l'intégrité des produits médicinaux lors de leur transport à travers la chaîne d'approvisionnement. Le PIB couvre des aspects tels que le stockage, le transport, la traçabilité et la gestion des risques de qualité. L'objectif est de garantir que les produits arrivent en toute sécurité, efficacement et intacte jusqu'aux consommateurs finaux.
GEP – Good Engineering Practice
Les bonnes pratiques d'ingénierie comprennent un ensemble de normes et de procédures appliquées dans la planification, la conception, la mise en œuvre et la maintenance des systèmes et projets d'ingénierie. Ces pratiques visent à garantir que toutes les solutions techniques et installations fonctionnent efficacement, en toute sécurité et en conformité avec les exigences réglementaires.
GLP – Good Laboratory Practice
Les bonnes pratiques de laboratoire sont un ensemble de règlements qui spécifient les exigences pour l'organisation, la planification et la conduite d'études non cliniques impliquant des substances et des produits pharmaceutiques. Il inclut également la documentation des résultats et le contrôle qualité de ces tests.
GMP – Good Manufacturing Practice / cGMP – current Good Manufacturing Practice
GMP – Good Manufacturing Practice, et sa version plus récente, cGMP – current Good Manufacturing Practice, décrivent des directives et réglementations visant à garantir une qualité de produit pharmaceutique et active de manière constante. Ces pratiques englobent divers aspects de la production, tels que la gestion de la qualité, les processus de fabrication et la maintenance des installations, afin d'assurer la sécurité, l'efficacité et le respect des normes réglementaires.
GSP – Good Storage Practices
Les bonnes pratiques de stockage impliquent des normes et des directives visant à stocker les produits de manière sûre, efficace et de manière à préserver leur qualité. Ces pratiques garantissent que les conditions de stockage, telles que la température et l'humidité, sont correctement contrôlées afin de préserver l'intégrité et l'efficacité des produits.
GxP/ cGxP
GxP signifie « Good x Practice » et est un terme général désignant diverses directives et réglementations de qualité appliquées dans des industries réglementées telles que les secteurs pharmaceutique, alimentaire et des dispositifs médicaux. Le « x » dans GxP peut être remplacé par différentes lettres pour couvrir différentes zones :
- GLP : Bonnes pratiques de laboratoire – liées à la qualité et à l'intégrité des résultats des données de laboratoire.
- GMP : Bonnes pratiques de fabrication – liées à l'assurance qualité pendant la production.
- PIB : Bonnes pratiques de distribution – désigne le stockage et le transport appropriés des produits.
- GCP : Good Clinical Practice – lié aux normes éthiques et scientifiques pour les essais cliniques.
- GEP : Good Engineering Practice – concerne les pratiques établies dans les projets techniques.
HACCP – Hazard Analysis and Critical Control Points
Le HACCP est un système proactif de gestion des risques conçu pour garantir la sécurité des produits alimentaires et pharmaceutiques. Le système HACCP identifie les dangers potentiels dans un processus de production et établit des points de contrôle critiques où des actions peuvent être prises pour minimiser ou éliminer ces dangers.
HAZOP – Hazard and Operability Study
Cette méthode est une approche systématique permettant d'identifier les risques et vulnérabilités potentiels dans les procédés industriels, en particulier dans les industries chimiques et pharmaceutiques. Les études HAZOP sont menées pour détecter des écarts par rapport au mode opérationnel prévu pouvant entraîner des dangers, et pour évaluer leur impact sur la sécurité, l'efficacité et la qualité. La procédure consiste en une analyse structurée des processus à l'aide de « mots guides » pour identifier les points de danger potentiels et les conditions de fonctionnement défaillantes.
ICH – International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use
L'ICH, à l'origine appelée « Conférence internationale sur l'harmonisation », a été créée en 1990 par la FDA, la Commission européenne, le ministère japonais de la Santé et les principales associations de l'industrie pharmaceutique. Basé à Genève, le ICH élabore des lignes directrices standardisées pour la qualité, l'efficacité et la sécurité des produits pharmaceutiques, tels que le GCP ou le GMP. Ces directives servent de directives à l'industrie pharmaceutique. Parmi les observateurs figurent l'OMS (Organisation mondiale de la santé) et l'AELE (Association européenne de libre-échange). Dans l'UE, ces directives sont adoptées par l'EMA (Agence européenne des médicaments) et sont considérées comme des normes dont les entreprises pharmaceutiques ne devraient s'écarter que dans des cas exceptionnels.
ICH Q9 – Quality Risk Management – Scientific Guideline
Ce document fournit des principes et des exemples de gestion des risques qualité qui peuvent être appliqués à divers aspects des mesures d'assurance qualité. Cela inclut le développement, la fabrication, la distribution, ainsi que les processus d'inspection et de soumission/examen tout au long du cycle de vie des substances actives, des produits pharmaceutiques, ainsi que des produits biologiques et biotechnologiques.
En résumé, le document contient des lignes directrices et des outils pour la gestion des risques de qualité applicables à l'ensemble de la chaîne de valeur des produits pharmaceutiques.
IQ – Installation Qualification
Cette phase du processus de validation est menée pour garantir que les équipements, systèmes ou installations sont correctement installés et respectent toutes les spécifications techniques, exigences du fabricant et normes réglementaires. Cela inclut, entre autres, l'acceptation d'une installation électrique appropriée, l'examen détaillé de tous les signaux d'entrée et de sortie, ainsi que le calcul correct de la valeur mesurée.
ISPE® – International Society for Pharmaceutical Engineering
ISPE est une organisation à but non lucratif qui promeut les avancées scientifiques, techniques et réglementaires dans l'industrie pharmaceutique. Il soutient ses membres par l'échange de connaissances, la formation et des ressources liées à l'ingénierie et aux processus réglementés, et participe à l'élaboration de normes et de réglementations.
ISPE® GAMP® 5 – A Risk-Based Approach to Compliant GxP Computerized Systems
GAMP® 5 (Good Automated Manufacturing Practice) est un guide utilisé pour la mise en œuvre et la validation de systèmes informatisés dans l'industrie pharmaceutique. Elle propose une approche basée sur les risques afin de garantir que tous les systèmes automatisés fonctionnent en conformité avec les exigences réglementaires applicables et garantit la qualité du produit et l'intégrité des données souhaitées. GAMP® 5 souligne l'importance d'une approche du cycle de vie pour la validation des systèmes, en tenant compte de la planification du projet, de la spécification, de la vérification et de la maintenance.
JP – Japanese Pharmacopeia
La Pharmacopée japonaise (JP) fournit des normes pour la qualité, la sécurité et l'efficacité des produits pharmaceutiques au Japon. Elle contient des normes obligatoires pour la fabrication, les tests et l'utilisation des produits pharmaceutiques.
LVP – Large Volume Parenteral
Les Parenterals à grand volume (LVP) sont des produits pharmaceutiques stériles emballés dans des contenants de plus de 100 ml. Ces produits sont principalement utilisés pour l'administration intraveineuse de liquides contenant des électrolytes, des nutriments ou des médicaments. Les LVP subissent des processus de stérilisation rigoureux, ou filtration aseptique, afin d'assurer leur sécurité et leur efficacité.
OOE – Out-of-Expectation
Les résultats « Out of Expectation » (OOE) sont des résultats qui s'avèrent être des anomalies singulières et, statistiquement parlant, n'ont pas de signification significative. Elles n'indiquent généralement pas un problème systémique. Les résultats de l'OOE sont généralement atypiques ou divergents qui ne correspondent pas aux autres données existantes. Contrairement à un résultat « hors de spécification » (OOS), les résultats OOE ne violent pas les limites établies de la spécification.
OOS – Out-of-Specification
Le terme « hors spécification » (OOS) désigne les résultats de tests qui ne correspondent pas aux critères d'acceptation établis, tels que définis dans les pharmacopées officielles ou les documents spécifiques à l'entreprise. Le terme est principalement utilisé dans le contexte des réglementations de la FDA. Les résultats OOS sont des résultats de tests qui ne respectent pas les normes spécifiées. Traiter ces écarts est une partie essentielle des systèmes d'assurance qualité tels que l'ISO 9001, les BPF et les BPL.
OOT – Out-of-Trend
L'OOT décrit un résultat qui se situe dans les limites spécifiées mais présente une tendance perceptible dans l'évaluation statistique. Ces résultats sont souvent appelés « hors de tendance » (OOT). Un résultat OOT indique que, bien que les résultats soient encore dans les spécifications établies, il existe une tendance ou un changement au fil du temps qui nécessite une attention particulière. De telles tendances peuvent être importantes pour identifier rapidement les problèmes potentiels et garantir la stabilité des processus.
OQ – Operational Qualification
L'OQ, ou Qualification Opérationnelle, est une étape cruciale dans le processus de validation des équipements et systèmes au sein des industries réglementées, telles que les secteurs pharmaceutique et des technologies médicales. Il vérifie et documente que tous les équipements, systèmes ou processus fonctionnent correctement dans leur environnement prévu et sur toute leur plage opérationnelle. Pendant la phase OQ, des protocoles et procédures de test spécifiques sont menés afin de garantir que tous les paramètres opérationnels respectent les spécifications établies.
PAT – Process Analytical Technology
Dans les industries pharmaceutique et biotechnologique, la technologie analytique de procédé (PAT) désigne un système permettant d'analyser et de contrôler les processus de fabrication par des mesures en temps réel des attributs critiques de qualité (CQA) du produit. L'objectif de PAT est d'obtenir une meilleure compréhension et un meilleur contrôle des procédés de fabrication afin d'optimiser la qualité des produits tout en réduisant les coûts et les délais de production.
PCS – Process Control System
Un système de contrôle de procédé est un système intégré de technologies et de procédures utilisé pour surveiller et contrôler les processus de production. Il garantit que ces processus fonctionnent efficacement, en toute sécurité et dans les paramètres spécifiés. Dans la fabrication pharmaceutique, un PCS est utilisé pour garantir que les produits intermédiaires et finis, tels que les ingrédients pharmaceutiques actifs (API), respectent les spécifications et les normes de qualité.
PDA® Parenteral Drug Association
L'Association des Médicaments Parentéraux (PDA®) est une organisation internationale axée sur l'avancement de la science et de la réglementation dans les industries pharmaceutique et biotechnologique. Elle soutient les professionnels de ces domaines en fournissant une formation, une expertise et des directives pour améliorer la qualité et la sécurité des médicaments parentéraux (c'est-à-dire administrés par voie orale) et des produits associés. La PDA® est bien connue pour son rôle dans l'élaboration des meilleures pratiques et des normes. Ses rapports techniques offrent des conseils pratiques pour un large éventail d'applications et sont souvent cités dans des normes et réglementations.
PHA – Preliminary Hazard Analysis
Cette méthode est une forme précoce d'analyse des risques utilisée lors de la phase de planification d'un projet ou lors de l'introduction de nouveaux procédés et produits. L'objectif de l'analyse préliminaire des dangers (PHA) est d'identifier les dangers potentiels et d'évaluer les risques avant la finalisation de la conception. L'analyse prend en compte les événements possibles pouvant entraîner des dangers, leurs impacts et les mesures de réduction des risques possibles.
PIC/S® – Pharmaceutical Inspection Cooperation Scheme
Le Programme de coopération en matière d'inspection pharmaceutique (PIC/S®) est un programme international de coopération initialement créé comme successeur de la Convention sur l'inspection pharmaceutique. L'objectif du PIC/S® est de promouvoir la coopération et l'harmonisation entre les autorités de supervision des Bonnes Pratiques de Fabrication (BPF) dans le monde entier. Cela est réalisé en standardisant les processus d'inspection et en améliorant la communication entre les organismes de régulation et l'industrie pharmaceutique. En publiant des directives et recommandations, le PIC/S® contribue à garantir la fabrication et la sécurité des produits pharmaceutiques à l'échelle mondiale. Les normes complètes fournies par le PIC/S® contribuent à améliorer l'assurance qualité et à éviter les inspections dites multiples.
PPQ – Process Performance Qualification
La qualification de performance de procédé (PPQ) est une étape cruciale de la validation de procédé, en particulier dans la deuxième phase appelée qualification de procédé. Elle va au-delà de la qualification de performance en évaluant la performance à long terme et la robustesse du processus de fabrication. Selon le GAMP® 5, le PPQ élargit la qualification d'une installation de production de produits médicaux et pharmaceutiques après l'achèvement de la qualification de performance.
PQ – Performance Qualification
La qualification de performance (PQ) est la vérification documentée que les installations, systèmes et équipements fonctionnent efficacement et avec des performances répétables conformément aux exigences approuvées. Cela inclut des tests dans des conditions réelles, la confirmation de la conformité aux normes de qualité, l'évaluation de la reproductibilité des résultats, ainsi qu'une documentation et une analyse minutieuses des données.
PQR – Product Quality Review
La Revue de la Qualité des Produits (PQR) est une évaluation périodique obligatoire par la loi qui garantit que les produits pharmaceutiques sont constamment fabriqués et contrôlés conformément aux exigences cGMP en matière de qualité, de sécurité et d'efficacité. En plus de l'analyse rétrospective, le PQR assure également le contrôle en temps réel des processus et l'amélioration continue en fournissant une vue d'ensemble complète de toutes les données pertinentes. En appliquant des outils statistiques, les écarts sont prédits et les tendances identifiées, permettant d'adopter des mesures correctives et préventives si nécessaire. La fréquence de la réalisation de l'examen dépend, entre autres facteurs, des résultats historiques de la revue, mais doit être réalisée au moins une fois par an.
Predictive Maintenance
Cette méthode utilise des technologies avancées pour la surveillance en temps réel et l'analyse des données afin de prédire quand la maintenance est nécessaire. Cela permet de détecter d'éventuelles défaillances et d'effectuer la maintenance uniquement lorsque cela est nécessaire, prolongeant la durée de vie des équipements et réduisant les temps d'arrêt ainsi que les coûts de maintenance. Un exemple de cela est constitué par les systèmes de surveillance continue tels que le système Kaye LabWatch.
Preventive Maintenance
La maintenance préventive consiste en des activités de maintenance planifiées qui sont réalisées quel que soit l'état de l'équipement. L'objectif est d'éviter les pannes et de prolonger la durée de vie des actifs. Cette approche proactive permet de garantir que les équipements fonctionnent efficacement et de manière fiable, réduisant ainsi les temps d'arrêt imprévus et les réparations potentiellement coûteuses.
Prospective Validation or Premarket Validation
La validation prospective consiste en des activités de validation menées avant qu'un produit ne soit fabriqué et mis en vente de façon routinière. L'objectif est de s'assurer qu'un procédé ou un système respecte les normes requises de qualité, de sécurité et d'efficacité avant une utilisation commerciale. Ce processus comprend la planification, l'exécution et la documentation des tests afin de se conformer aux exigences réglementaires.
PS – Purified Steam
La vapeur purifiée est celle qui répond aux critères stricts de pureté pour être considérée comme Eau à Injection (WFI) une fois condensée. Dans l'industrie pharmaceutique, la vapeur propre est principalement utilisée pour la stérilisation des composants entrant en contact avec des produits. De plus, il est utilisé pour humidifier les réserves d'air dans les salles blanches et les isolateurs afin d'assurer un environnement contrôlé et stérile.
PV – Process Validation
La validation des processus est une procédure étape par étape et documentée visant à garantir que les processus, dans leurs paramètres de conception établis, sont capables de produire de manière cohérente un produit final de la qualité requise. L'étendue de la validation est largement déterminée par une analyse des risques réalisée précédemment. Plus le risque est grand, plus l'effort de validation requis est élevé.
PW – Purified Water
L'eau purifiée est celle qui a été libérée des impuretés chimiques et microbiologiques par divers procédés, tels que la distillation, l'osmose inverse ou l'échange d'ions. Il est principalement utilisé dans l'industrie pharmaceutique, où des exigences de pureté élevées existent mais où la stérilité ou les conditions sans pyrogènes ne sont pas nécessaires. L'eau purifiée est utilisée dans la fabrication de produits pharmaceutiques non injectables, comme solvant ou comme base pour la production de produits comme les solutions de dialyse, à condition qu'elle réponde à certaines exigences de pureté, telles que le test d'endotoxine selon les normes pharmacopéiennes.
QA – Quality Assurance
L'assurance qualité (QA) est un processus systématique axé sur la surveillance et l'amélioration des normes de qualité dans la fabrication (axée sur les procédés) et la livraison de produits ou services. Dans l'industrie pharmaceutique, l'assurance qualité garantit que tous les produits respectent les normes de qualité et les exigences réglementaires requises, de la vérification de la qualité des matières premières à l'examen complet des produits finaux.
QbD – Quality-by-Design
L'Agence européenne des médicaments (EMA) soutient l'application de la qualité par conception (QbD) pour optimiser la fabrication pharmaceutique. Cette approche garantit la qualité des produits pharmaceutiques grâce à des méthodes statistiques et analytiques ainsi qu'à la gestion des risques. Il s'agit d'identifier et de contrôler les sources de variabilité afin de s'assurer que les médicaments respectent des caractéristiques prédéfinies dès le départ. Le concept utilise des analyses multivariées et des outils modernes pour mieux comprendre les attributs critiques et les paramètres de production, permettant des améliorations continues.
QC – Quality Control
Le contrôle qualité (QC) est un processus systématique dans l'industrie manufacturière visant à garantir que les produits (axés sur le produit) respectent les normes de qualité établies. La QC consiste à tester et inspecter les produits à différents stades de la production afin de s'assurer qu'ils respectent les spécifications et sont exempts de défauts. Ce processus inclut l'identification et la correction des défauts, ainsi que la vérification de la conformité aux réglementations et normes.
QMS – Quality Management System
Un système de gestion de la qualité (QMS) dans le cadre des bonnes pratiques de fabrication (BPF) est un système structuré de procédures, de procédés et de ressources conçu pour garantir la qualité et la sécurité des produits pharmaceutiques tout au long du processus de production. Elle englobe tous les aspects de la gestion de la qualité, y compris la planification, le contrôle, l'assurance qualité et l'amélioration continue, afin de se conformer aux exigences réglementaires et d'assurer l'efficacité et la sécurité des médicaments.
QP – Qualification Protocol
Un protocole de qualification (QP) est un plan ou une procédure écrite qui détaille la manière dont la qualification doit être obtenue. Il inclut des exigences de qualification spécifiques pour chaque équipement, chaque exigence système et exigence produit.
QP – Qualified Person
La Personne Qualifiée en pharmacie joue un rôle clé dans le droit pharmaceutique européen, responsable de la fabrication, des tests et de la mise en circulation des produits médicinaux conformément à la réglementation. Ils garantissent également une documentation complète du respect des exigences légales.
QSM – Quality System Manual
Un Manuel du Système de Qualité est un document complet qui décrit la structure, les processus et les politiques du Système de Gestion de la Qualité (QMS) d'une entreprise. Il établit les normes et procédures conçues pour garantir que les produits et services respectent les exigences de qualité requises. Le manuel sert de guide pour tous les employés afin d'atteindre les objectifs de qualité de l'entreprise et d'assurer le respect des exigences légales et spécifiques à chaque secteur.
Qualification
Le terme « qualification » dans l'environnement GxP fait référence au processus de vérification de l'adéquation d'un dispositif, d'une installation ou d'un système à son objectif prévu. La première mention de « qualification » dans la réglementation GxP est difficile à identifier, mais elle remonte à la fin des années 1970, lorsque la FDA a introduit la validation dans le cadre des bonnes pratiques de fabrication (GMP). Ce processus comprend les qualifications de conception, d'installation, d'exploitation et de performance (DQ, IQ, OQ, PQ). Le processus exact de qualification peut varier selon le contexte et l'appareil ou le système spécifique. La qualification est divisée en étapes exécutables individuelles. Il constitue une composante de la validation et concerne les performances et caractéristiques des systèmes, équipements, instruments, installations et logiciels. Il comprend généralement les étapes suivantes :
- URS (User Requirement Specification) : document qui capture et définit les exigences pour un système ou un produit spécifique.
- DQ (Qualification de conception) : Processus de vérification qui confirme que la conception de l'équipement ou du système répond à toutes les exigences prédéfinies pour l'objectif prévu.
- FAT/SAT (Test d'acceptation en usine/test d'acceptation sur site) : Le FAT est un processus de test avant expédition effectué sur le site du fabricant pour confirmer la conception et la conformité opérationnelle du système. Le SAT valide l'installation correcte et la performance fonctionnelle du système sur le site du client avant le début complet de l'exploitation.
- IQ (Qualification d'installation) : Vérification que l'équipement ou le système est correctement installé et fonctionne selon les paramètres prévus dans l'environnement prévu.
- OQ (Qualification opérationnelle) : Vérification que l'équipement ou le système fonctionne comme prévu sur toute la zone opérationnelle dans son environnement spécifique.
- PQ (Performance Qualification) : Confirmation et documentation que l'équipement ou le système peut fonctionner de manière cohérente selon les paramètres définis dans les URS (User Requirement Specifications)
Retrospective Validation
La validation rétrospective consiste à revoir et analyser les données de production existantes et les enregistrements de lots afin d'évaluer la cohérence et la performance d'un procédé déjà utilisé. L'objectif est de déterminer si le procédé a constamment produit des produits avec les attributs de qualité souhaités et s'il respecte aux normes de qualité et réglementaires basées sur des données historiques.
Revalidation
La revalidation est le processus de réévaluation et de répétition des activités de validation afin de s'assurer que les systèmes, processus ou équipements continuent de respecter les normes établies de qualité et de performance. Il consiste à analyser les données de performance existantes et est crucial pour maintenir le statut validé, en particulier lorsque des changements importants surviennent ou pour garantir une conformité continue aux exigences réglementaires.
Risk Acceptance
L'acceptation du risque fait référence à la volonté d'accepter le risque, que ce soit de manière formelle ou passive. Même avec de bonnes pratiques de gestion des risques, tous les risques ne peuvent pas être éliminés. Ainsi, le risque est souvent réduit à un niveau spécifié et acceptable, déterminé en fonction de la situation.
Risk Analysis
L'analyse des risques est l'évaluation des risques associés aux dangers identifiés. C'est le processus qualitatif ou quantitatif qui relie la probabilité de survenue à la gravité du dommage potentiel. Dans certains outils de gestion des risques, la capacité à détecter le dommage (détectabilité) joue également un rôle dans l'évaluation du risque.
Risk Assessment
Évaluation de la survenue d'un risque identifié et évaluation de sa probabilité et de son impact. Questions générales à considérer :
- Qu'est-ce qui pourrait mal tourner ?
- Quelle est la probabilité que cela tourne mal ?
- Quelles sont les conséquences (gravité) ?
Risk Communication
Échange d'informations sur les risques et la gestion des risques entre tous les départements impliqués dans le processus, y compris le contrôle qualité, la gestion de la qualité et la direction exécutive.
Risk Control
Établir des critères d'acceptation du risque, de réduction du risque et d'acceptation du risque. Le contrôle des risques consiste à prendre des décisions pour réduire et/ou accepter les risques. L'objectif du contrôle des risques est de réduire le risque à un niveau acceptable. L'effort de contrôle des risques doit être proportionnel à l'importance du risque.
Le contrôle des risques pourrait se concentrer sur les questions suivantes :
- Le risque est-il supérieur à un niveau acceptable ?
- Que peut-on faire pour réduire ou éliminer les risques ?
- Quel est l'équilibre approprié entre bénéfices, risques et ressources ?
- De nouveaux risques sont-ils introduits au fur et à mesure que les risques identifiés sont contrôlés ?
Risk Evaluation
L'évaluation des risques compare les risques identifiés et analysés avec des critères de risque établis.
Risk Identification
Identifier, capturer et documenter les risques potentiels pouvant impacter la qualité et la sécurité est un élément essentiel d'une gestion efficace des risques. Ce processus consiste à évaluer systématiquement les sources possibles de risque afin de mettre en œuvre des stratégies d'atténuation appropriées.
Risk Management
La gestion des risques est un élément central de l'environnement GxP tout au long du cycle de vie d'une installation/processus/système/instrument et fait partie des directives et réglementations. Le processus de gestion des risques vise à garantir que tous les risques sont identifiés, évalués et suffisamment contrôlés afin de garantir la qualité et la sécurité des produits pharmaceutiques. Les cinq étapes fondamentales de la gestion des risques sont :
- Identification des risques
- Évaluation des risques
- Contrôle des risques
- Communication des risques
- Examen des risques
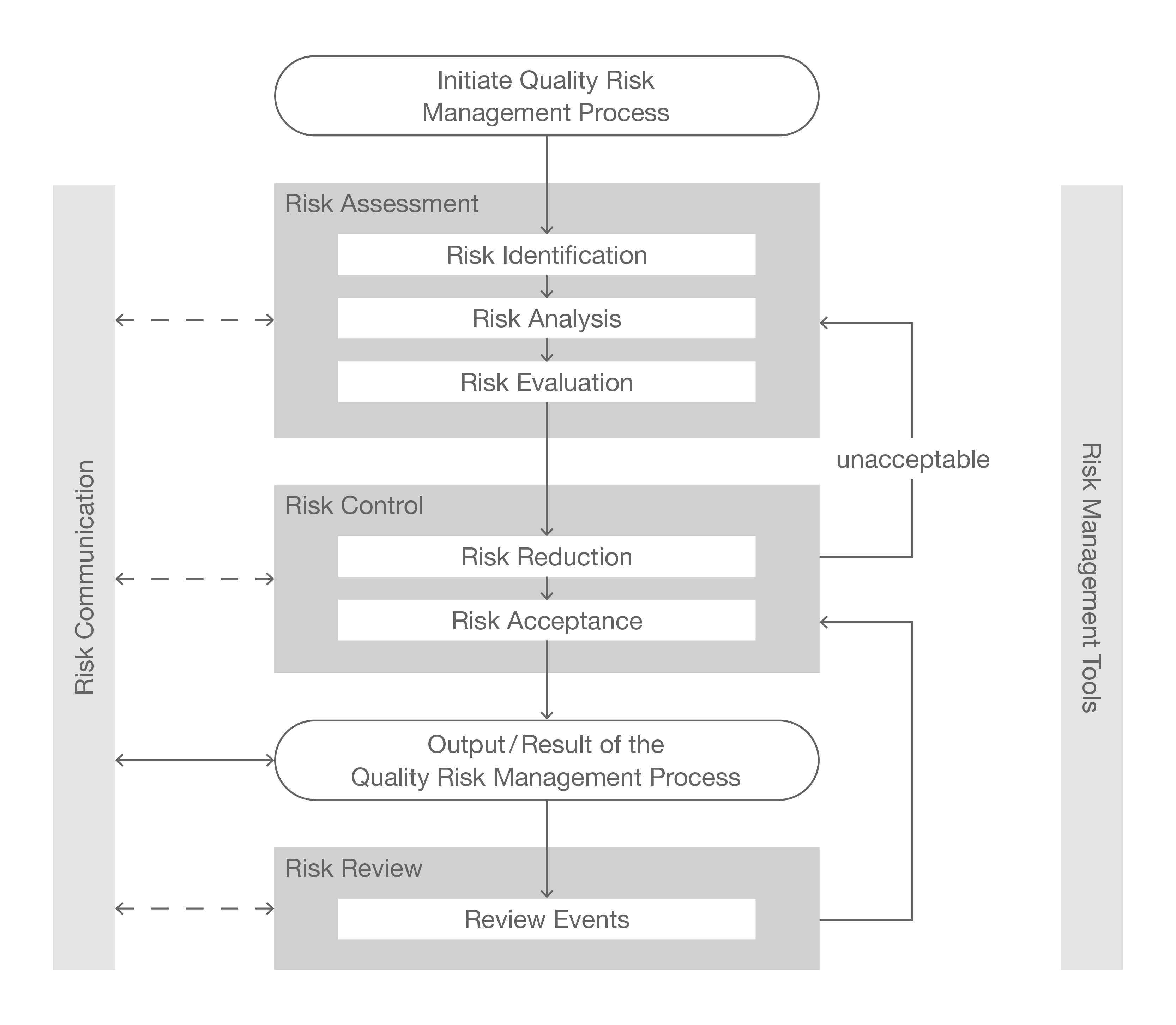
Risk Management Tools
La gestion des risques de qualité fournit des méthodes scientifiques et pratiques pour la prise de décision en gestion des risques, basées sur l'évaluation de la probabilité, de la gravité et de la détectabilité des risques. Les outils traditionnels et modernes de gestion des risques peuvent être appliqués en combinaison pour offrir de la flexibilité. Ces méthodes sont adaptées à la complexité et à la critique des questions à traiter. Une gamme d'outils tels que FMEA, FTA, HACCP, PHA et HAZOP soutiennent ce processus.
Risk Reduction
La réduction des risques consiste à agir pour diminuer ou éviter les risques de qualité lorsqu'ils dépassent un niveau acceptable. Bien que les mesures aident à réduire la gravité et la probabilité de dommages et à améliorer la détectabilité, elles peuvent aussi introduire de nouveaux risques ou influencer ceux existants. Par conséquent, l'évaluation des risques doit être examinée après la mise en œuvre des mesures d'atténuation des risques.
Risk Review
Fait référence à la révision et à l'évaluation régulières du processus de gestion des risques et de ses résultats afin de garantir qu'il reste à jour et efficace. La gestion des risques doit être intégrée en continu dans le processus de gestion de la qualité, avec des mécanismes de suivi et d'évaluation continue. Les résultats doivent être régulièrement révisés et ajustés pour intégrer de nouvelles connaissances. Les modifications, telles que les inspections programmées ou les rappels non planifiés, doivent être intégrées au processus en fonction des résultats de l'examen des risques.
SAL – Sterility Assurance Level
Le niveau d'assurance de la stérilité, également appelé SAL, décrit la probabilité qu'un seul produit reste contaminé après le processus de stérilisation. Cette métrique est principalement utilisée dans la fabrication de produits pharmaceutiques stériles et de dispositifs médicaux.
Sanitization
La désinfection est un processus en plusieurs étapes et peut généralement être divisée en plusieurs étapes communes :
- Préparation : Élimination des matériaux inutiles et fourniture d'agents de nettoyage et de désinfection.
- Nettoyage : Élimination complète de la saleté et des matières organiques des surfaces.
- Rinçage : Élimination des résidus d'agent nettoyant par un rinçage approfondi.
- Désinfection : Application de désinfectants pour tuer les micro-organismes restants ; dans la désinfection à froid, des agents chimiques sont utilisés à basse température.
- Inspection et documentation : Vérification du processus de désinfection et documentation de toutes les mesures visant à garantir les normes d'hygiène.
Les différences entre la désinfection à chaud et à froid résident dans la plage de températures et la méthode de réduction des micro-organismes :
- La désinfection à chaud utilise des températures comprises entre 80 et 90°C, comme de l'eau chaude ou de la vapeur, pour tuer les bactéries, virus et champignons, mais elle est énergivore et peut provoquer l'usure des matériaux.
- La désinfection à froid utilise des agents chimiques à basse température, idéal pour les matériaux sensibles à la température, mais nécessite une sélection et une manipulation soigneuses des produits
SAT – Site-Acceptance-Test
Un test d'acceptation de site (SAT) est un processus critique qui garantit qu'une machine ou un système est correctement installé, configuré et opérationnel sur le site du client. Il évalue si l'équipement répond aux exigences opérationnelles et documente l'état actuel afin d'enregistrer les écarts par rapport aux spécifications. Le processus comprend la vérification de l'installation, de la fonctionnalité des composants, de la conformité aux normes de sécurité et des tests de performance pour garantir la qualité et la sécurité avant le début de l'exploitation régulière.
SIP – Steam in Place
La stérilisation à la vapeur en (SIP) est un procédé crucial dans l'industrie pharmaceutique pour garantir la stérilité des systèmes installés de façon permanente tels que les réservoirs, les pipelines et les bioréacteurs dans la fabrication aseptique. Ce procédé utilise de la vapeur saturée pour tuer les micro-organismes sans nécessiter de démonter l'équipement. Les mêmes normes et réglementations appliquées aux autoclaves à vapeur/stérilisateurs à vapeur sont utilisées dans le SIP. Les normes pertinentes pour la stérilisation à la chaleur humide, telles que l'EN 285 ou l'ISO 17665, sont prises en compte. En général, le processus CIP (Clean-In-Place) précède le processus SIP, préparant l'équipement à la stérilisation.
SMF – Site Master File
Un Site Master File (SMF) est une partie centrale de la documentation dans la fabrication pharmaceutique, contenant des informations sur un site de production et ses installations. Il est créé par le fabricant pharmaceutique pour garantir la conformité aux bonnes pratiques de fabrication (BPF) et aux normes réglementaires. Le SMF comprend les détails du site, les qualifications du personnel, les processus de production, les systèmes de contrôle qualité, les conditions de distribution et de transport, ainsi que les certifications. Il sert de document essentiel pour les autorités réglementaires, les évaluations clients et les inspections.
SOP – Standard Operation Procedure
Des instructions de travail documentées détaillant les processus et tâches régulièrement exécutés. Ces instructions servent à garantir la cohérence et la qualité dans l'exécution des processus. Les procédures opérationnelles standard (SOP) sont essentielles pour garantir que tous les employés respectent les normes et réglementations requises, assurant ainsi la qualité des produits, la sécurité et la conformité réglementaire.
Steam Quality
En stérilisation à la vapeur, la qualité de la vapeur fait référence aux propriétés spécifiques de la vapeur nécessaires à une stérilisation réussie. Il définit le rapport masse de la vapeur à l'eau dans un mélange vapeur-eau.
Les propriétés de la vapeur suivantes sont importantes pour la stérilisation :
- Une forte teneur énergétique pour un transfert de chaleur optimal et l'atteinte des températures de stérilisation.
- Capacité de pénétration accrue pour assurer une couverture complète des objets à stériliser.
- Capacité à condenser et à déplacer l'air de la chambre, facilitant une stérilisation efficace.
Une mauvaise qualité de vapeur peut nuire à la stérilisation, car le transfert de chaleur et la pénétration de la vapeur peuvent être entravés. Par conséquent, la surveillance et le contrôle de la qualité de la vapeur lors de la stérilisation à la chaleur humide sont essentiels.
SVPs – Small Volume Parenteral
Les Small Volume Parenteral (SVP) désignent des solutions d'injection stériles généralement emballées en volumes inférieurs à 100 ml. Ces solutions sont emballées dans différents contenants selon leur usage prévu. Les SVP sont couramment utilisés pour l'administration de médicaments et doivent respecter des exigences strictes de stérilisation.
Temperature (Heat) Penetration Study
L'objectif d'une étude de pénétration de température est de déterminer les caractéristiques de chauffage et de refroidissement d'une combinaison produit/emballage dans un procédé de stérilisation spécifique afin d'établir des procédés thermiques sûrs et d'évaluer les écarts de procédé. L'étude doit être conçue pour examiner de manière adéquate et précise tous les facteurs critiques affectant les taux de chauffage associés au produit, à l'emballage et au procédé. Les études de pénétration sont souvent menées en parallèle avec les études de distribution de température.
Temperature Mapping Study
Les études de répartition des températures sont appliquées dans divers domaines de la gestion des GMP ainsi que dans différentes directives et normes. Ils sont réalisés là où la température est cruciale pour la qualité du procédé et, en fin de compte, pour la qualité du produit final. Cela inclut les processus de stérilisation, les systèmes de refroidissement et de congélation, les salles de stockage, les chambres de stabilité et les conteneurs de transport au sein de la chaîne du froid. Par exemple, dans un autoclave à vapeur, le point froid et le point chaud sont déterminés, tandis que la cartographie de température dans une salle de stockage permet, entre autres, de placer des capteurs de surveillance fixes.
Traceability
La traçabilité signifie généralement pouvoir suivre toutes les étapes d'un produit ou d'un procédé d'un lot ainsi que les actions menées depuis l'origine des matières premières jusqu'à la distribution finale du produit fini. Par exemple, la traçabilité dans l'étalonnage d'un capteur de température signifie que l'étalonnage et l'ajustement peuvent être retracés jusqu'à une norme nationale de température.
URS – User-Requirements-Specifications
Il s'agit d'un document qui décrit les exigences et attentes du client pour un nouveau produit ou système. Il définit ce que le système doit accomplir et, avec les exigences techniques et réglementaires, constitue la base du document de spécifications. Le document de spécifications est généralement créé par le client ou le client.
Ce document est un composant du V-Modèle. Il regroupe les exigences provenant de diverses sources et soutient la conception, la mise en service, la qualification, ainsi que l'exploitation et la maintenance d'un système. Les attributs critiques de qualité et les paramètres du processus sont des composants importants à identifier pour soutenir le processus basé sur le risque de qualité. Les spécifications peuvent être conçues de manière flexible pour répondre aux besoins d'une opération polyvalente et sont des documents dynamiques qui sont mis à jour lorsque des changements surviennent.
USP® – United States Pharmacopeia
La Pharmacopée des États-Unis (USP®) est une organisation américaine à but non lucratif qui élabore des normes pour l'assurance qualité des médicaments et compléments alimentaires. Ces normes sont importantes pour la conformité réglementaire dans l'industrie pharmaceutique mondiale.
Validation
La validation est le processus documenté de vérification qu'un système, un procédé, un équipement ou un produit répond de manière fiable aux exigences spécifiques et aux normes de qualité. Ce procédé garantit que tous les paramètres critiques sont contrôlés et produisent systématiquement des résultats reproductibles afin de garantir à la fois la sécurité et l'efficacité du produit final. Ainsi, la validation est la preuve documentée que toutes les procédures, processus, installations, systèmes, matériaux et équipements respectent les exigences GxP et fournissent les résultats attendus.
Validation Matrix
En particulier dans les validations complexes, la matrice de validation facilite la surveillance. Il s'agit d'un document structuré ou d'un outil qui représente la relation entre diverses exigences de validation et les éléments à valider, tels que les systèmes, processus ou équipements. Cette matrice permet de garantir que tous les aspects d'un projet respectent les normes réglementaires et internes de l'entreprise.
Typiquement, une matrice de validation inclut des informations sur :
- Exigences de validation : Critères spécifiques à remplir en fonction des directives réglementaires et des normes de l'entreprise.
- Éléments à valider : Liste des systèmes, processus ou composants à valider.
- Tests et critères : Méthodes et procédures appliquées pour confirmer le respect des exigences.
- Documentation : Références aux documents contenant les résultats et preuves de la validation.
- Responsabilités : Attribution de tâches à des individus ou équipes spécifiques pour la réalisation d'activités de validation.
Validation Plan
vérifié qu'un système ou un produit répond aux exigences établies. Pour y parvenir, des tests et inspections spécifiques sont planifiés et documentés. En plus de la planification, une analyse des risques est souvent nécessaire pour identifier les faiblesses potentielles et développer des mesures de validation ciblées afin d'atténuer les risques.
Dans de nombreux secteurs, en particulier dans la pharmacie, la médecine et l'alimentation, le respect des exigences réglementaires oriente la planification de la validation. Il est crucial que tous les tests et inspections respectent ces réglementations pour garantir leur conformité. Une documentation approfondie du processus de validation est essentielle pour démontrer le respect des exigences de qualité, notamment lors des audits ou inspections.
Validation Protocol
Un protocole de validation est un résumé détaillé et écrit qui décrit des tests et procédures spécifiques permettant de vérifier si un produit répond aux exigences de son objectif prévu. Il inclut des instructions pour la validation des critères d'acceptation et la documentation nécessaire des résultats. Le protocole est plus ciblé que le plan de validation et se concentre sur la mise en œuvre pratique de la validation pour des processus ou produits spécifiques.
Validation Report
Après l'achèvement des activités de validation, le rapport de validation documente les résultats et évalue si les exigences de validation ont été remplies. Le rapport inclut l'analyse et l'interprétation des données, indique si les critères d'acceptation ont été atteints et propose des recommandations pour les actions futures.
VMP – Validation Master Plan
Le Plan Directeur de Validation (VMP) est un document stratégique qui guide toutes les activités de validation d'une usine de fabrication afin d'assurer la qualité constante des produits et la conformité aux normes de qualité. En tant qu'élément essentiel de l'engagement qualité d'une entreprise, le VMP garantit que les produits et procédés sont systématiquement validés. Il soutient la cohérence et le contrôle de la production, minimise les risques et assure la conformité réglementaire grâce à une documentation détaillée et à l'alignement avec les réglementations en vigueur. De plus, le VMP optimise l'utilisation des ressources et fournit des instructions claires pour la minimisation des erreurs. Elle doit se conformer aux exigences réglementaires applicables, telles que celles de la FDA et de l'UE. En général, le VMP établit la stratégie de validation de l'entreprise et définit et documente les intentions, responsabilités, approches et aspects clés d'un programme de validation.
WFI – Water-for-Injection
L'eau pour injection (WFI) est une méthode d'eau hautement purifiée, spécifiquement utilisée dans la production de produits pharmaceutiques pour l'administration parentérale. Il sert de matériau porteur pour dissoudre ou diluer des substances ou préparations destinées à être injectées dans l'organisme. Le WFI doit répondre à des exigences strictes de qualité afin de garantir qu'il est exempt d'impuretés et de contamination microbiologique. Dans l'industrie pharmaceutique, il s'agit d'un élément essentiel pour garantir la sécurité et l'efficacité des préparations injectables.
WHO – World Health Organization
L'Organisation mondiale de la santé (OMS), fondée en 1948 et dont le siège est à Genève, sert d'autorité de coordination pour les questions internationales de santé publique. Son objectif est de créer un avenir meilleur et plus sain pour les populations du monde entier.
WIP – Washing-in-Place
WIP (Washing In Place) désigne le nettoyage et la désinfection semi-automatisés, par exemple, des machines de remplissage en injectant des agents de nettoyage et de désinfection appropriés dans le système. Contrairement au CIP (Clean-In-Place), le WIP nécessite un effort de nettoyage manuel supplémentaire.
Z-Value
La valeur Z est un paramètre microbiologique qui caractérise le comportement de destruction des micro-organismes lors de la stérilisation. Il décrit l'augmentation nécessaire de la température pour réduire la valeur D d'un facteur dix, ce qui entraîne une réduction en logarithèmes des micro-organismes. La valeur Z indique la sensibilité des microbes aux variations de température et est essentielle pour garantir une réduction efficace des micro-organismes avec un impact minimal sur le produit.
ZLG – Zentralstelle der Länder für Gesundheitsschutz bei Arzneimitteln und Medizinprodukten
La Zentralstelle der Länder für Gesundheitsschutz bei Arzneimitteln und Medizinprodukten (ZLG) est une institution allemande qui coordonne la coopération entre les États fédéraux sur les questions de protection de la santé. Elle joue un rôle important dans l'harmonisation et le suivi des normes pour les produits médicinaux et les dispositifs médicaux en Allemagne. Le ZLG soutient les autorités étatiques dans la surveillance, l'approbation et la certification des produits et procédés dans le domaine médical afin d'assurer la sécurité et l'efficacité. Grâce à cette approche coordonnée, le ZLG contribue à garantir des normes de qualité élevées en matière de protection de la santé.